





Abstract
The human genome is a mosaic with respect to its evolutionary history. Based on a phylogenetic analysis of 23,210 DNA sequence alignments from human, chimpanzee, gorilla, orangutan, and rhesus, we present a map of human genetic ancestry. For about 23% of our genome, we share no immediate genetic ancestry with our closest living relative, the chimpanzee. This encompasses genes and exons to the same extent as intergenic regions. We conclude that about 1/3 of our genes started to evolve as human-specific lineages before the differentiation of human, chimps, and gorillas took place. This explains recurrent findings of very old human-specific morphological traits in the fossils record, which predate the recent emergence of the human species about 5-6 MYA. Furthermore, the sorting of such ancestral phenotypic polymorphisms in subsequent speciation events provides a parsimonious explanation why evolutionary derived characteristics are shared among species that are not each other's closest relatives.
Introduction
Reconstructing the evolutionary process that has molded contemporary humans out of the ancestors shared with their closest relatives, the great apes, is one of the key objectives in evolutionary research (Nuttal 1904; Goodman 1962; King and Wilson 1975; Schwartz 1984; Sibley and Ahlquist 1984; Horai et al. 1992; Shoshani et al. 1996; Ruvolo 1997; Satta et al. 2000; Klein and Takahata 2002).
However, with both amount of data and number of studies increasing, the crux of the matter emerges. Regardless of the type of phylogenetically informative data chosen for analysis, the evolutionary history of humans is reconstructed differently with different sets of data (summarized in Ebersberger 2004). The dilemma is best exemplified when genetic distances are considered for evolutionary studies. The extent of DNA sequence divergence between humans and chimpanzees in combination with various calibration points for the molecular clock places the split of the 2 species around 6 million years before present (MYBP) (Glazko and Nei 2003; Patterson et al. 2006). This considerably young age of the human species conflicts with at least part of the fossil record on early human evolution (Brunet et al. 2005; Patterson et al. 2006). Moreover, datings obtained from different regions of the human genome vary over a range of more than 4 Myr—some placing the human-chimp split as recent as 4 MYBP (Patterson et al. 2006)—where exons seem to support older splits than introns (Osada and Wu 2005). Eventually, when genetic distances are considered to infer the evolutionary relationships among humans and the great apes, even the species genetically most similar to humans varies with the locus under study (Ruvolo 1997; Satta et al. 2000; Chen and Li 2001; Patterson et al. 2006; Hobolth et al. 2007).
To understand why regions in the human genome can differ in their evolutionary history, it needs to be acknowledged that genetic lineages represented by DNA sequences in the extant species trace back to allelic variants in the shared ancestral species (Nei 1987) (fig. 1). In here, these variants persist until they join in their most recent common ancestor (MRCA). Some genetic lineages, however, do not coalesce in the progenitor exclusively shared by humans and chimpanzees. They enter, together with the lineage descending from the gorilla, the ancestral population of all 3 species, where any 2 of the 3 lineages can merge first. Thus, in two-thirds of the cases, a genealogy results in which humans and chimpanzees are not each other's closest genetic relatives. The corresponding genealogies are incongruent with the species tree. In concordance with the experimental evidences, this implies that there is no such thing as a unique evolutionary history of the human genome. Rather, it resembles a patchwork of individual regions following their own genealogy.
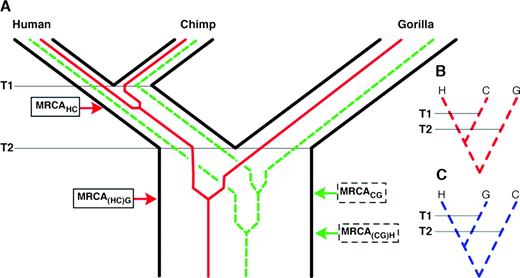
(A) The distinct evolutionary histories of species and their genes. The species tree is drawn in black. T1 and T2 denote the speciation events of humans and chimpanzees, and of gorillas, respectively. Human and chimpanzee genetic lineages can coalesce in their MRCAHC in the progenitor exclusively shared by the 2 species (red solid sequence tree). Under a model of random genetic drift, this occurs with p(H,C) = e−T(HC)/(2Ne × g), where T(HC) is the time span between T1 and T2, Ne is the effective population size of the ancestral species, and g is its generation time. The branching pattern of the resulting sequence tree is congruent to the species tree. For the dashed sequence trees, human and chimpanzee genetic lineages fail to coalesce in the exclusive ancestor of both species. In the ancestral species shared by humans, chimpanzees, and gorillas any 2 of the 3 lineages can join first. The green graph depicts the coalescent event resulting in a common ancestry of chimpanzees and gorillas (MRCACG). The remaining 2 branching patterns, (H,C)G and (H,G)C, are shown in (B) and (C), respectively.
The recent availability of a chimpanzee genome draft sequence and its comparison to the human genome has resulted in a genome-wide collection of genetic differences between the 2 species (The Chimpanzee Sequencing and Analysis Consortium 2005). A subset of these differences forms the genetic background for the specific phenotypic characteristics of humans (Vigilant and Paabo 1999; Varki 2000; Ebersberger 2004). Accordingly, this catalogue of genetic differences has been referred to as the ultimate resource to study human and chimpanzee biology and evolution (Li and Saunders 2005). It is now interesting to determine when in the history of the 2 species and on what lineage these evolutionary changes have occurred (Chou et al. 2002; Enard et al. 2002; Gilad et al. 2003). From this perspective, particularly the issue that a fraction of our genome does not have the chimpanzee as our closest relative gains new momentum. Neither do we know for a particular human DNA sequence the genetic sister species nor do we know when the corresponding genetic lineages have separated. However, only an answer to both questions will help to track down the genetic changes that formed contemporary humans and chimpanzees.
In the present study, we focus on 3 aspects. First, what is the fraction of the human genome, and particularly of the genes therein, for which chimpanzees are not our closest relatives, and how are these regions distributed along human chromosomes. To this end, we compare a genome-wide collection of 23,210 human and chimpanzee DNA sequences with their homologs in gorilla, orangutan, and rhesus, respectively. Based on a likelihood approach, we first identify those sequence trees that significantly reject chimpanzees as our closest relatives, that is, sequence trees that are incongruent with the species tree. Second, we reestimate the splitting times for the human and great ape lineages and assess the ancestral population sizes of the ancient species shared by humans and chimpanzees from the fraction of incongruent sequence trees. Third, we determine the position of incongruent sequence trees relative to genes and exons in the human genome and discuss the consequences of the complex genetic ancestry of humans for our view on human and chimpanzee evolution.
Materials and Methods
Data
We downloaded 33,018 alignments of DNA sequences from human, chimpanzee, gorilla, orangutan, and rhesus, originating from a large-scale shotgun sample sequencing study of a western lowland gorilla (HCGOM data set [Patterson et al. 2006]) from http://genepath.med.harvard.edu/∼reich.
Data Preprocessing
The HCGOM data set is available only as Arachne alignments (Jaffe et al. 2003) of DNA shotgun sequence reads from the 4 nonhuman primate species to the National Center for Biotechnology Information (NCBI) Build 34 human genome assembly. No information on the quality of the individual aligned sequence reads was provided. To utilize this data set nonetheless, a number of preprocessing steps were required. For each Arachne alignment, we first extracted the individual sequence reads from the gorilla and the orangutan, respectively. Sequences from the chimpanzee and rhesus were ignored at this step because we used the assembled genome sequences instead. For each sequence, we retrieved the corresponding source sequence and its quality information from the NCBI trace archive (http://www.ncbi.nlm.nih.gov/Traces). This step excluded 6,459 Arachne alignments from further analysis because for at least one orangutan sequence we could find no entry in the NCBI trace archive. We next identified start and end of the subsequence that was used in the Arachne alignment in the source sequence. With this positional information, the corresponding base quality substring was extracted. Subsequently, we assembled the gorilla and orangutan sequences separately into contigs using Phrap (http://www.phrap.org). These contigs were then aligned with the human genome sequence (NCBI Build 36) using BLAT (Kent 2002) with setting “minScore = 300.” If more than one BLAT hit was obtained, the one with the highest score was retained. Human genome sequence positions covered by both gorilla and orangutan sequences were then identified and the corresponding human DNA sequence was extracted with nibFrag (http://www.soe.ucsc.edu/∼kent/src/unzipped/utils/nibFrag/) from the Build 36 genome assembly. To obtain the orthologous regions in the chimpanzee and rhesus genomes, we used the pairwise human–chimpanzee and human–rhesus genome assemblies provided at the Human Genome Browser Web site (Kent et al. 2002). The corresponding DNA sequences together with their base quality information were then extracted from the chimpanzee genome assembly (PanTro2) and the rhesus genome assembly (rheMac2). Subsequently, we aligned the DNA sequences from the 5 primate species with ClustalW (Thompson et al. 1994). From these multiple sequence alignment, we eventually extracted those regions where sequences from all 5 species overlap. The resulting 30,112 multiple sequence alignments are available upon request.
Quality Screen
Positions with a Phred value below 20 were masked in the aligned chimpanzee, gorilla, orangutan, and rhesus sequences, respectively (Ewing and Green 1998; Taudien et al. 2006). Increasing the threshold to a Phred value of 40 had no significant effect on the outcome of the analysis but a mere decrease of the number of analyzed position. Alignment regions with a clustering of masked positions are indicative of an overall low sequence quality. To identify and remove such regions, we penalized alignment columns including a masked position with a minimum of −2 (one masked nucleotide) and a maximum of −6 (all nonhuman nucleotides have been masked) and rewarding unmasked columns with +1. Then we extracted the highest scoring subalignment. Only subalignments with a length ≥300 bp were further analyzed, leaving 26,909 alignments.
Phylogenetic Analysis
Phylogenetic tree reconstructions and molecular clock tests were performed with Tree-Puzzle (Schmidt et al. 2002) using the Hasegawa-Kishino-Yano model of DNA sequence evolution (Hasegawa et al. 1985) and 4 rate categories to model substitution rate heterogeneity among sites. Alignments for which the automatic root search implemented into Tree-Puzzle did not place the root on the rhesus branch were removed from the analysis (leaving 25,500 alignments), as where such alignments for which the molecular clock was rejected on a significance level of 0.05 (leaving 23,210 alignments). Posterior probabilities for the 15 unrooted tree topologies were estimated from the individual likelihoods assuming a uniform prior distribution on the set of trees. The fraction of incongruent sequence trees was assumed to follow a binomial distribution. Ninety-five percent confidence intervals (CI) were assessed using the normal approximation to the binomial distribution.
Dating of Speciation Events
To assess the splitting times of the 5 species, we first inferred the sequence tree together with its branch lengths from a concatenation of the alignments that did not reject the molecular clock and for which the root was placed on the rhesus branch. However, splitting times estimated from DNA sequence comparisons predate the historical separation of the species. To minimize this bias, we omitted alignments where the inferred sequence tree deviates significantly (posterior probability ≥ 0.95) from the species tree. The split of the orangutan 16 MYBP was used to calibrate the molecular clock.
Identification of Sequence Alignments Overlapping with Human Genes
To identify alignments that overlap with genes and exons in the human genome, we compared the alignment position with the position of “known genes” in the human genome as annotated in the Ensembl release 39 (http://www.ensembl.org/Homo_sapiens/index.html).
Analysis of Gene Ontology Terms
To investigate whether gene function has an influence on the phylogeny of the gene sequence, we analyzed the representation of Gene Ontology (GO) terms in our data. To this end, we used FatiGO2 (Al-Shahrour et al. 2006) to screen for GO terms that are overrepresented in the fraction of genes for which at least one exon overlaps with an alignment that significantly supports an incongruent sequence tree. As a reference, we used those genes in our data set to which a sequence tree was mapped that does not differ significantly from the species tree.
Results
The finished human genome sequence (International Human Genome Sequencing Consortium 2004) has been recently complemented by draft versions of the chimpanzee and rhesus genomes (The Chimpanzee Sequencing and Analysis Consortium 2005); (http://www.hgsc.bcm.tmc.edu/projects/macaque) and extensive amounts of whole-genome shotgun sequences from the orangutan (ftp://ftp.ncbi.nih.gov/pub/TraceDB/pongo_pygmaeus). In a recent study on the evolutionary origin of humans, this data set has been further extended by 115,152 shotgun sequences determined from randomly chosen regions of the gorilla genome (Patterson et al. 2006). A combination of the sequence information from the 5 species (chimpanzee, human, gorilla, orangutan, and rhesus) leads to more than 30,000 Arachne (Jaffe et al. 2003) alignments (Patterson et al. 2006). This data are used to address the question of our genetic ancestry on a genome-wide scale with a likelihood-based approach. Processing of the Arachne alignments (see Materials and Methods) provided a total of 23,210 clocklike evolving DNA sequence alignments of 5 species each, summing up to a total of 14,512,620 compared nucleotide positions (table 1).
Data Per Human Chromosome
| Chromosome | Unresolved Treesa (bp) | Congruent Trees (bp) | Incongruent Trees (bp) | Incongruent Trees !(HC)b (bp) | P!(HC)c | (95% CI) | THCd |
| 1 | 875 (534,348) | 746 (489,475) | 222 (141,003) | 216 (137,014) | 0.225 | (0.199–0.252) | 2.09 |
| 2 | 1083 (662,878) | 784 (503,638) | 258 (164,281) | 250 (158,893) | 0.242 | (0.217–0.269) | 2.05 |
| 3 | 844 (511,478) | 689 (439,967) | 224 (143,461) | 216 (138,385) | 0.239 | (0.212–0.268) | 2.04 |
| 4 | 800 (485,332) | 623 (401,151) | 240 (152,474) | 234 (148,818) | 0.273 | (0.244–0.304) | 2.17 |
| 5 | 793 (482,433) | 593 (384,869) | 164 (103,521) | 163 (102,841) | 0.216 | (0.188–0.246) | 2.02 |
| 6 | 681 (413,617) | 562 (362,229) | 166 (108,306) | 162 (105,457) | 0.224 | (0.195–0.256) | 2.09 |
| 7 | 622 (376,931) | 494 (322,539) | 122 (79,787) | 116 (75,690) | 0.190 | (0.161–0.223) | 2.07 |
| 8 | 652 (389,957) | 489 (309,131) | 206 (133,763) | 203 (131,967) | 0.293* | (0.261–0.328) | 2.01 |
| 9 | 430 (263,814) | 321 (206,528) | 95 (62,077) | 94 (61,431) | 0.227 | (0.189–0.269) | 2.10 |
| 10 | 532 (321,819) | 451 (297,923) | 129 (79,779) | 126 (78,550) | 0.218 | (0.187–0.254) | 2.07 |
| 11 | 527 (323,719) | 427 (271,289) | 139 (90,011) | 135 (87,587) | 0.240 | (0.207–0.277) | 2.11 |
| 12 | 534 (316,150) | 434 (279,528) | 126 (79,077) | 123 (77,411) | 0.221 | (0.188–0.257) | 2.13 |
| 13 | 413 (247,541) | 339 (218,931) | 100 (62,176) | 100 (62,176) | 0.228 | (0.191–0.269) | 2.15 |
| 14 | 335 (205,718) | 278 (179,268) | 106 (67,427) | 104 (66,273) | 0.272 | (0.230–0.319) | 2.11 |
| 15 | 352 (212,254) | 264 (170,824) | 87 (57,416) | 83 (54,871) | 0.239 | (0.197–0.287) | 2.01 |
| 16 | 294 (177,154) | 220 (142,967) | 53 (33,605) | 53 (33,605) | 0.194 | (0.152–0.245) | 1.98 |
| 17 | 254 (151,447) | 204 (130,653) | 61 (40,574) | 59 (39,189) | 0.224 | (0.178–0.279) | 1.99 |
| 18 | 333 (203,240) | 279 (181,758) | 95 (60,631) | 94 (60,187) | 0.252 | (0.211–0.298) | 2.18 |
| 19 | 140 (83,264) | 79 (50,123) | 28 (17,064) | 26 (15,821) | 0.248 | (0.175–0.338) | 1.96 |
| 20 | 213 (130,284) | 225 (142,255) | 63 (41,026) | 62 (40,480) | 0.216 | (0.172–0.267) | 2.25 |
| 21 | 122 (75,791) | 101 (63,346) | 40 (24,768) | 40 (24,768) | 0.284 | (0.216–0.363) | 2.07 |
| 22 | 83 (47,862) | 90 (57,120) | 21 (13,672) | 21 (13,672) | 0.189 | (0.127–0.272) | 2.11 |
| X | 354 (207,153) | 455 (294,019) | 52 (33,006) | 50 (31,988) | 0.099* | (0.076–0.128) | 2.82 |
| Total | 11,266 (6,824,184) | 9,147 (5,899,531) | 2,797 (1,788,905) | 2,730 (1,747,074) | 0.230 | (0.222–0.238) | 2.08 |
| Chromosome | Unresolved Treesa (bp) | Congruent Trees (bp) | Incongruent Trees (bp) | Incongruent Trees !(HC)b (bp) | P!(HC)c | (95% CI) | THCd |
| 1 | 875 (534,348) | 746 (489,475) | 222 (141,003) | 216 (137,014) | 0.225 | (0.199–0.252) | 2.09 |
| 2 | 1083 (662,878) | 784 (503,638) | 258 (164,281) | 250 (158,893) | 0.242 | (0.217–0.269) | 2.05 |
| 3 | 844 (511,478) | 689 (439,967) | 224 (143,461) | 216 (138,385) | 0.239 | (0.212–0.268) | 2.04 |
| 4 | 800 (485,332) | 623 (401,151) | 240 (152,474) | 234 (148,818) | 0.273 | (0.244–0.304) | 2.17 |
| 5 | 793 (482,433) | 593 (384,869) | 164 (103,521) | 163 (102,841) | 0.216 | (0.188–0.246) | 2.02 |
| 6 | 681 (413,617) | 562 (362,229) | 166 (108,306) | 162 (105,457) | 0.224 | (0.195–0.256) | 2.09 |
| 7 | 622 (376,931) | 494 (322,539) | 122 (79,787) | 116 (75,690) | 0.190 | (0.161–0.223) | 2.07 |
| 8 | 652 (389,957) | 489 (309,131) | 206 (133,763) | 203 (131,967) | 0.293* | (0.261–0.328) | 2.01 |
| 9 | 430 (263,814) | 321 (206,528) | 95 (62,077) | 94 (61,431) | 0.227 | (0.189–0.269) | 2.10 |
| 10 | 532 (321,819) | 451 (297,923) | 129 (79,779) | 126 (78,550) | 0.218 | (0.187–0.254) | 2.07 |
| 11 | 527 (323,719) | 427 (271,289) | 139 (90,011) | 135 (87,587) | 0.240 | (0.207–0.277) | 2.11 |
| 12 | 534 (316,150) | 434 (279,528) | 126 (79,077) | 123 (77,411) | 0.221 | (0.188–0.257) | 2.13 |
| 13 | 413 (247,541) | 339 (218,931) | 100 (62,176) | 100 (62,176) | 0.228 | (0.191–0.269) | 2.15 |
| 14 | 335 (205,718) | 278 (179,268) | 106 (67,427) | 104 (66,273) | 0.272 | (0.230–0.319) | 2.11 |
| 15 | 352 (212,254) | 264 (170,824) | 87 (57,416) | 83 (54,871) | 0.239 | (0.197–0.287) | 2.01 |
| 16 | 294 (177,154) | 220 (142,967) | 53 (33,605) | 53 (33,605) | 0.194 | (0.152–0.245) | 1.98 |
| 17 | 254 (151,447) | 204 (130,653) | 61 (40,574) | 59 (39,189) | 0.224 | (0.178–0.279) | 1.99 |
| 18 | 333 (203,240) | 279 (181,758) | 95 (60,631) | 94 (60,187) | 0.252 | (0.211–0.298) | 2.18 |
| 19 | 140 (83,264) | 79 (50,123) | 28 (17,064) | 26 (15,821) | 0.248 | (0.175–0.338) | 1.96 |
| 20 | 213 (130,284) | 225 (142,255) | 63 (41,026) | 62 (40,480) | 0.216 | (0.172–0.267) | 2.25 |
| 21 | 122 (75,791) | 101 (63,346) | 40 (24,768) | 40 (24,768) | 0.284 | (0.216–0.363) | 2.07 |
| 22 | 83 (47,862) | 90 (57,120) | 21 (13,672) | 21 (13,672) | 0.189 | (0.127–0.272) | 2.11 |
| X | 354 (207,153) | 455 (294,019) | 52 (33,006) | 50 (31,988) | 0.099* | (0.076–0.128) | 2.82 |
| Total | 11,266 (6,824,184) | 9,147 (5,899,531) | 2,797 (1,788,905) | 2,730 (1,747,074) | 0.230 | (0.222–0.238) | 2.08 |
The maximum likelihood tree has a posterior probability below 0.95.
Incongruent sequence trees that differ from the species tree only in the branching order of humans, chimpanzees, and gorillas (!(HC)).
Probability to observe a !(HC) tree and the corresponding 95% CI. Chromosomes that differ significantly from the genome-wide average of 0.230 are marked with an asterisk.
Estimated time span the shared ancestral species to humans and chimpanzees has existed in million years. For this column, “Total” represents the estimate obtained from the concatenation of only the autosomal sequences.
Data Per Human Chromosome
| Chromosome | Unresolved Treesa (bp) | Congruent Trees (bp) | Incongruent Trees (bp) | Incongruent Trees !(HC)b (bp) | P!(HC)c | (95% CI) | THCd |
| 1 | 875 (534,348) | 746 (489,475) | 222 (141,003) | 216 (137,014) | 0.225 | (0.199–0.252) | 2.09 |
| 2 | 1083 (662,878) | 784 (503,638) | 258 (164,281) | 250 (158,893) | 0.242 | (0.217–0.269) | 2.05 |
| 3 | 844 (511,478) | 689 (439,967) | 224 (143,461) | 216 (138,385) | 0.239 | (0.212–0.268) | 2.04 |
| 4 | 800 (485,332) | 623 (401,151) | 240 (152,474) | 234 (148,818) | 0.273 | (0.244–0.304) | 2.17 |
| 5 | 793 (482,433) | 593 (384,869) | 164 (103,521) | 163 (102,841) | 0.216 | (0.188–0.246) | 2.02 |
| 6 | 681 (413,617) | 562 (362,229) | 166 (108,306) | 162 (105,457) | 0.224 | (0.195–0.256) | 2.09 |
| 7 | 622 (376,931) | 494 (322,539) | 122 (79,787) | 116 (75,690) | 0.190 | (0.161–0.223) | 2.07 |
| 8 | 652 (389,957) | 489 (309,131) | 206 (133,763) | 203 (131,967) | 0.293* | (0.261–0.328) | 2.01 |
| 9 | 430 (263,814) | 321 (206,528) | 95 (62,077) | 94 (61,431) | 0.227 | (0.189–0.269) | 2.10 |
| 10 | 532 (321,819) | 451 (297,923) | 129 (79,779) | 126 (78,550) | 0.218 | (0.187–0.254) | 2.07 |
| 11 | 527 (323,719) | 427 (271,289) | 139 (90,011) | 135 (87,587) | 0.240 | (0.207–0.277) | 2.11 |
| 12 | 534 (316,150) | 434 (279,528) | 126 (79,077) | 123 (77,411) | 0.221 | (0.188–0.257) | 2.13 |
| 13 | 413 (247,541) | 339 (218,931) | 100 (62,176) | 100 (62,176) | 0.228 | (0.191–0.269) | 2.15 |
| 14 | 335 (205,718) | 278 (179,268) | 106 (67,427) | 104 (66,273) | 0.272 | (0.230–0.319) | 2.11 |
| 15 | 352 (212,254) | 264 (170,824) | 87 (57,416) | 83 (54,871) | 0.239 | (0.197–0.287) | 2.01 |
| 16 | 294 (177,154) | 220 (142,967) | 53 (33,605) | 53 (33,605) | 0.194 | (0.152–0.245) | 1.98 |
| 17 | 254 (151,447) | 204 (130,653) | 61 (40,574) | 59 (39,189) | 0.224 | (0.178–0.279) | 1.99 |
| 18 | 333 (203,240) | 279 (181,758) | 95 (60,631) | 94 (60,187) | 0.252 | (0.211–0.298) | 2.18 |
| 19 | 140 (83,264) | 79 (50,123) | 28 (17,064) | 26 (15,821) | 0.248 | (0.175–0.338) | 1.96 |
| 20 | 213 (130,284) | 225 (142,255) | 63 (41,026) | 62 (40,480) | 0.216 | (0.172–0.267) | 2.25 |
| 21 | 122 (75,791) | 101 (63,346) | 40 (24,768) | 40 (24,768) | 0.284 | (0.216–0.363) | 2.07 |
| 22 | 83 (47,862) | 90 (57,120) | 21 (13,672) | 21 (13,672) | 0.189 | (0.127–0.272) | 2.11 |
| X | 354 (207,153) | 455 (294,019) | 52 (33,006) | 50 (31,988) | 0.099* | (0.076–0.128) | 2.82 |
| Total | 11,266 (6,824,184) | 9,147 (5,899,531) | 2,797 (1,788,905) | 2,730 (1,747,074) | 0.230 | (0.222–0.238) | 2.08 |
| Chromosome | Unresolved Treesa (bp) | Congruent Trees (bp) | Incongruent Trees (bp) | Incongruent Trees !(HC)b (bp) | P!(HC)c | (95% CI) | THCd |
| 1 | 875 (534,348) | 746 (489,475) | 222 (141,003) | 216 (137,014) | 0.225 | (0.199–0.252) | 2.09 |
| 2 | 1083 (662,878) | 784 (503,638) | 258 (164,281) | 250 (158,893) | 0.242 | (0.217–0.269) | 2.05 |
| 3 | 844 (511,478) | 689 (439,967) | 224 (143,461) | 216 (138,385) | 0.239 | (0.212–0.268) | 2.04 |
| 4 | 800 (485,332) | 623 (401,151) | 240 (152,474) | 234 (148,818) | 0.273 | (0.244–0.304) | 2.17 |
| 5 | 793 (482,433) | 593 (384,869) | 164 (103,521) | 163 (102,841) | 0.216 | (0.188–0.246) | 2.02 |
| 6 | 681 (413,617) | 562 (362,229) | 166 (108,306) | 162 (105,457) | 0.224 | (0.195–0.256) | 2.09 |
| 7 | 622 (376,931) | 494 (322,539) | 122 (79,787) | 116 (75,690) | 0.190 | (0.161–0.223) | 2.07 |
| 8 | 652 (389,957) | 489 (309,131) | 206 (133,763) | 203 (131,967) | 0.293* | (0.261–0.328) | 2.01 |
| 9 | 430 (263,814) | 321 (206,528) | 95 (62,077) | 94 (61,431) | 0.227 | (0.189–0.269) | 2.10 |
| 10 | 532 (321,819) | 451 (297,923) | 129 (79,779) | 126 (78,550) | 0.218 | (0.187–0.254) | 2.07 |
| 11 | 527 (323,719) | 427 (271,289) | 139 (90,011) | 135 (87,587) | 0.240 | (0.207–0.277) | 2.11 |
| 12 | 534 (316,150) | 434 (279,528) | 126 (79,077) | 123 (77,411) | 0.221 | (0.188–0.257) | 2.13 |
| 13 | 413 (247,541) | 339 (218,931) | 100 (62,176) | 100 (62,176) | 0.228 | (0.191–0.269) | 2.15 |
| 14 | 335 (205,718) | 278 (179,268) | 106 (67,427) | 104 (66,273) | 0.272 | (0.230–0.319) | 2.11 |
| 15 | 352 (212,254) | 264 (170,824) | 87 (57,416) | 83 (54,871) | 0.239 | (0.197–0.287) | 2.01 |
| 16 | 294 (177,154) | 220 (142,967) | 53 (33,605) | 53 (33,605) | 0.194 | (0.152–0.245) | 1.98 |
| 17 | 254 (151,447) | 204 (130,653) | 61 (40,574) | 59 (39,189) | 0.224 | (0.178–0.279) | 1.99 |
| 18 | 333 (203,240) | 279 (181,758) | 95 (60,631) | 94 (60,187) | 0.252 | (0.211–0.298) | 2.18 |
| 19 | 140 (83,264) | 79 (50,123) | 28 (17,064) | 26 (15,821) | 0.248 | (0.175–0.338) | 1.96 |
| 20 | 213 (130,284) | 225 (142,255) | 63 (41,026) | 62 (40,480) | 0.216 | (0.172–0.267) | 2.25 |
| 21 | 122 (75,791) | 101 (63,346) | 40 (24,768) | 40 (24,768) | 0.284 | (0.216–0.363) | 2.07 |
| 22 | 83 (47,862) | 90 (57,120) | 21 (13,672) | 21 (13,672) | 0.189 | (0.127–0.272) | 2.11 |
| X | 354 (207,153) | 455 (294,019) | 52 (33,006) | 50 (31,988) | 0.099* | (0.076–0.128) | 2.82 |
| Total | 11,266 (6,824,184) | 9,147 (5,899,531) | 2,797 (1,788,905) | 2,730 (1,747,074) | 0.230 | (0.222–0.238) | 2.08 |
The maximum likelihood tree has a posterior probability below 0.95.
Incongruent sequence trees that differ from the species tree only in the branching order of humans, chimpanzees, and gorillas (!(HC)).
Probability to observe a !(HC) tree and the corresponding 95% CI. Chromosomes that differ significantly from the genome-wide average of 0.230 are marked with an asterisk.
Estimated time span the shared ancestral species to humans and chimpanzees has existed in million years. For this column, “Total” represents the estimate obtained from the concatenation of only the autosomal sequences.
Contrasting Phylogenetic Signals in the Data
We first assessed the fraction of sequence trees that is incongruent with the species tree. To this end, we calculated the likelihoods for all 15 unrooted tree topologies (Felsenstein 1981) for each individual alignment. If we take the maximum likelihood tree at face value to represent the phylogeny of the alignment, then 9,343 (40%) of the sequence trees are incongruent with the species tree (table 2). Among these trees, the majority (8,630) places the orangutan basal to humans and the African apes. The incongruence with the species tree is restricted to the subtree connecting humans, chimps, and gorillas. The remaining 713 alignments do not recover the monophyly of humans, chimpanzees, and gorillas. Alas, the 5 species under study and thus their DNA sequences are in part very closely related. As pointed out (Takahata and Satta 1997; Yang 2002), this may result in an erroneous reconstruction of the sequence tree because different branching patterns have similar likelihood values. The likelihood-mapping plot (Strimmer and von Haeseler 1997) in figure 2 gives an impression about the extent of phylogenetic information in the data with respect to the resolution of the subtree connecting humans, chimpanzees, and gorillas. Figure 2C shows that ∼40% of the alignments provide no clear support for a single branching pattern. Consistent with the prediction that the inclusion of alignments with no clear phylogenetic signal leads to an overestimation of the fraction of incongruent sequence trees (Yang 2002), we observe an increasing proportion of incongruent sequence trees with decreasing phylogenetic information in our data (supplementary fig. 1, Supplementary Material online).
Number of Alignments in Support of the 15 Sequence Tree Topologies Featuring the Monophyly of the Great Apes
| Topology | All (%) | Genea (%) | Exonb (%) |
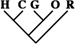 | 174 (0.75) | 70 (0.72) | 15 (1.06) |
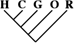 | 13,869 (59.75) | 5,869 (60.39) | 805 (57.05) |
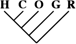 | 205 (0.88) | 101 (1.04) | 23 (1.63) |
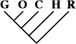 | 15 (0.06) | 5 (0.05) | 3 (0.21) |
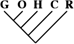 | 29 (0.12) | 14 (0.14) | 0 (0) |
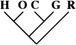 | 50 (0.22) | 17 (0.17) | 3 (0.21) |
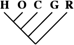 | 25 (0.11) | 12 (0.12) | 2 (0.14) |
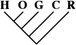 | 20 (0.09) | 7 (0.07) | 1 (0.07) |
 | 41 (0.18) | 19 (0.20) | 5 (0.35) |
 | 4,140 (17.84) | 1,650 (16.98) | 242 (17.15) |
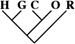 | 43 (0.19) | 23 (0.24) | 5 (0.35) |
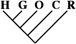 | 64 (0.28) | 26 (0.27) | 8 (0.57) |
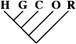 | 4,490 (19.34) | 1,881 (19.36) | 289 (20.48) |
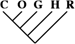 | 14 (0.06) | 5 (0.05) | 2 (0.14) |
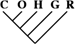 | 33 (0.14) | 19 (0.20) | 8 (0.57) |
| Topology | All (%) | Genea (%) | Exonb (%) |
| 174 (0.75) | 70 (0.72) | 15 (1.06) |
| 13,869 (59.75) | 5,869 (60.39) | 805 (57.05) |
| 205 (0.88) | 101 (1.04) | 23 (1.63) |
| 15 (0.06) | 5 (0.05) | 3 (0.21) |
| 29 (0.12) | 14 (0.14) | 0 (0) |
| 50 (0.22) | 17 (0.17) | 3 (0.21) |
| 25 (0.11) | 12 (0.12) | 2 (0.14) |
| 20 (0.09) | 7 (0.07) | 1 (0.07) |
| 41 (0.18) | 19 (0.20) | 5 (0.35) |
| 4,140 (17.84) | 1,650 (16.98) | 242 (17.15) |
| 43 (0.19) | 23 (0.24) | 5 (0.35) |
| 64 (0.28) | 26 (0.27) | 8 (0.57) |
| 4,490 (19.34) | 1,881 (19.36) | 289 (20.48) |
| 14 (0.06) | 5 (0.05) | 2 (0.14) |
| 33 (0.14) | 19 (0.20) | 8 (0.57) |
Alignments that overlap with the position of a gene in the human genome.
Alignments that overlap with the position of an exon in the human genome.
Number of Alignments in Support of the 15 Sequence Tree Topologies Featuring the Monophyly of the Great Apes
| Topology | All (%) | Genea (%) | Exonb (%) |
| 174 (0.75) | 70 (0.72) | 15 (1.06) |
| 13,869 (59.75) | 5,869 (60.39) | 805 (57.05) |
| 205 (0.88) | 101 (1.04) | 23 (1.63) |
| 15 (0.06) | 5 (0.05) | 3 (0.21) |
| 29 (0.12) | 14 (0.14) | 0 (0) |
| 50 (0.22) | 17 (0.17) | 3 (0.21) |
| 25 (0.11) | 12 (0.12) | 2 (0.14) |
| 20 (0.09) | 7 (0.07) | 1 (0.07) |
| 41 (0.18) | 19 (0.20) | 5 (0.35) |
| 4,140 (17.84) | 1,650 (16.98) | 242 (17.15) |
| 43 (0.19) | 23 (0.24) | 5 (0.35) |
| 64 (0.28) | 26 (0.27) | 8 (0.57) |
| 4,490 (19.34) | 1,881 (19.36) | 289 (20.48) |
| 14 (0.06) | 5 (0.05) | 2 (0.14) |
| 33 (0.14) | 19 (0.20) | 8 (0.57) |
| Topology | All (%) | Genea (%) | Exonb (%) |
| 174 (0.75) | 70 (0.72) | 15 (1.06) |
| 13,869 (59.75) | 5,869 (60.39) | 805 (57.05) |
| 205 (0.88) | 101 (1.04) | 23 (1.63) |
| 15 (0.06) | 5 (0.05) | 3 (0.21) |
| 29 (0.12) | 14 (0.14) | 0 (0) |
| 50 (0.22) | 17 (0.17) | 3 (0.21) |
| 25 (0.11) | 12 (0.12) | 2 (0.14) |
| 20 (0.09) | 7 (0.07) | 1 (0.07) |
| 41 (0.18) | 19 (0.20) | 5 (0.35) |
| 4,140 (17.84) | 1,650 (16.98) | 242 (17.15) |
| 43 (0.19) | 23 (0.24) | 5 (0.35) |
| 64 (0.28) | 26 (0.27) | 8 (0.57) |
| 4,490 (19.34) | 1,881 (19.36) | 289 (20.48) |
| 14 (0.06) | 5 (0.05) | 2 (0.14) |
| 33 (0.14) | 19 (0.20) | 8 (0.57) |
Alignments that overlap with the position of a gene in the human genome.
Alignments that overlap with the position of an exon in the human genome.
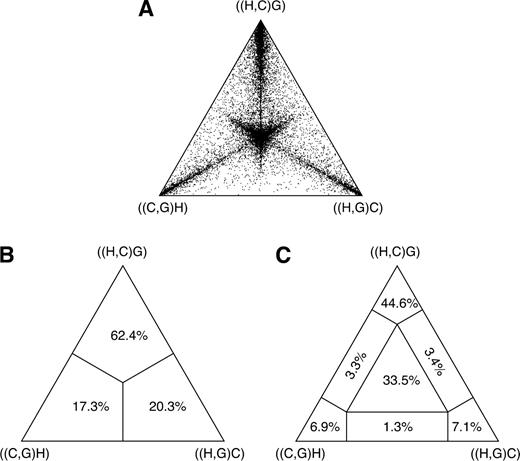
Likelihood-mapping plot of the 5-species alignments supporting the monophyly of humans and the great apes. (A) Each point in the equilateral triangle represents a single alignment. The lengths of the perpendiculars from any point to the triangle sides are equal to the posterior probabilities of each of the 3 topologies of the subtree connecting human, chimpanzee, and gorilla ((H,C)G; (H,G)C; (C,G)H) is supported by the corresponding alignment (Strimmer and von Haeseler 1997). The closer a point is located to 1 of the 3 edges, the higher the support for the respective phylogeny. (B) Fraction of the data for which the support for the respective sequence tree is largest. (C) The area of the triangle is separated into 7 regions reflecting different phylogenetic information contents of the data. Alignments located in the central area fail to resolve the human-chimpanzee-gorilla subtree. Alignments located in the corner areas support the corresponding sequence tree. The remaining 3 regions contain those alignments for which it is not possible to decide between 2 of the 3 topologies. Numbers represent the percentage of the data set located in the 7 areas.
To identify the subset of our data significantly supporting only a single phylogeny, we consider only sequence trees that are supported with a posterior probability of at least 95%. This leaves us with 11,945 phylogenetically informative alignments (tables 1 and 3). Among these, 23.0% (95% CI 22.2–23.8%) support a closer relationship of gorilla to either humans or chimpanzees, although they recover the monophyly of the 3 species. Trees where the gorilla is placed closer to the chimpanzee and trees with a human–gorilla sister group are observed equally often (1,369 and 1,361, respectively). Note that still 0.6% (95% CI 0.4–0.7%) of the resolved sequence trees place the orang within the human–chimp–gorilla subtree.
Number of Alignments Significantly (posterior probability ≥ 0.95) Supporting the 15 Sequence Tree Topologies Featuring the Monophyly of the Great Apes
| Topology | All (%) | Genea (%) | Exonb (%) |
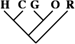 | 20 (0.17) | 8 (0.17) | 2 (0.32) |
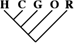 | 9,148 (76.58) | 3,814 (78.85) | 487 (78.93) |
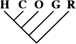 | 19 (0.16) | 10 (0.21) | 2 (0.32) |
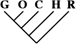 | 0 | 0 | 0 |
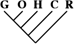 | 1 (0.01) | 0 | 0 |
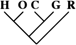 | 5 (0.04) | 2 (0.04) | 0 |
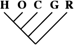 | 0 | 0 | 0 |
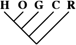 | 0 | 0 | 0 |
 | 4 (0.03) | 1 (0.02) | 0 |
 | 1,369 (11.46) | 504 (10.42) | 63 (10.21) |
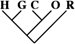 | 13 (0.11) | 6 (0.12) | 1 (0.16) |
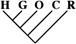 | 5 (0.04) | 0 | 0 |
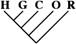 | 1,361 (11.39) | 492 (10.17) | 62 (10.05) |
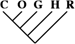 | 0 | 0 | 0 |
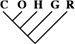 | 0 | 0 | 0 |
| Topology | All (%) | Genea (%) | Exonb (%) |
| 20 (0.17) | 8 (0.17) | 2 (0.32) |
| 9,148 (76.58) | 3,814 (78.85) | 487 (78.93) |
| 19 (0.16) | 10 (0.21) | 2 (0.32) |
| 0 | 0 | 0 |
| 1 (0.01) | 0 | 0 |
| 5 (0.04) | 2 (0.04) | 0 |
| 0 | 0 | 0 |
| 0 | 0 | 0 |
| 4 (0.03) | 1 (0.02) | 0 |
| 1,369 (11.46) | 504 (10.42) | 63 (10.21) |
| 13 (0.11) | 6 (0.12) | 1 (0.16) |
| 5 (0.04) | 0 | 0 |
| 1,361 (11.39) | 492 (10.17) | 62 (10.05) |
| 0 | 0 | 0 |
| 0 | 0 | 0 |
Alignments that overlap with the position of a gene in the human genome.
Alignments that overlap with the position of an exon in the human genome.
Number of Alignments Significantly (posterior probability ≥ 0.95) Supporting the 15 Sequence Tree Topologies Featuring the Monophyly of the Great Apes
| Topology | All (%) | Genea (%) | Exonb (%) |
| 20 (0.17) | 8 (0.17) | 2 (0.32) |
| 9,148 (76.58) | 3,814 (78.85) | 487 (78.93) |
| 19 (0.16) | 10 (0.21) | 2 (0.32) |
| 0 | 0 | 0 |
| 1 (0.01) | 0 | 0 |
| 5 (0.04) | 2 (0.04) | 0 |
| 0 | 0 | 0 |
| 0 | 0 | 0 |
| 4 (0.03) | 1 (0.02) | 0 |
| 1,369 (11.46) | 504 (10.42) | 63 (10.21) |
| 13 (0.11) | 6 (0.12) | 1 (0.16) |
| 5 (0.04) | 0 | 0 |
| 1,361 (11.39) | 492 (10.17) | 62 (10.05) |
| 0 | 0 | 0 |
| 0 | 0 | 0 |
| Topology | All (%) | Genea (%) | Exonb (%) |
| 20 (0.17) | 8 (0.17) | 2 (0.32) |
| 9,148 (76.58) | 3,814 (78.85) | 487 (78.93) |
| 19 (0.16) | 10 (0.21) | 2 (0.32) |
| 0 | 0 | 0 |
| 1 (0.01) | 0 | 0 |
| 5 (0.04) | 2 (0.04) | 0 |
| 0 | 0 | 0 |
| 0 | 0 | 0 |
| 4 (0.03) | 1 (0.02) | 0 |
| 1,369 (11.46) | 504 (10.42) | 63 (10.21) |
| 13 (0.11) | 6 (0.12) | 1 (0.16) |
| 5 (0.04) | 0 | 0 |
| 1,361 (11.39) | 492 (10.17) | 62 (10.05) |
| 0 | 0 | 0 |
| 0 | 0 | 0 |
Alignments that overlap with the position of a gene in the human genome.
Alignments that overlap with the position of an exon in the human genome.
Subsequently, we checked whether the amount of incongruent sequence trees varies in different subsets of the human genome. First, we considered alignments that overlap with genes and exons. No significant difference in the frequency of the individual tree topologies compared with the genome-wide average is seen (table 3). This figure changes when we assess the fraction of incongruent sequence trees separately for the individual chromosomes (table 1 and fig. 3A). Values range between a low of 9.9% (95% CI 7.6–12.8%) for human chromosome X to a high of 29.3% (95% CI 26.1–32.8%) for human chromosome 8. A 1-factorial analysis of variance and a subsequent Bonferroni posttest rejected the hypothesis that all autosomes show the same mean fraction of incongruent sequence trees (P < 0.01). However, in a subsequent pairwise comparison, only chromosomes 7 and 8, displaying the smallest and largest proportion of incongruent trees, respectively, differ significantly. Including the X chromosome into the analysis revealed that the fraction of incongruent sequence trees on the sex chromosome is significantly different to that observed on all human autosomes, except for chromosome 7 and 22.
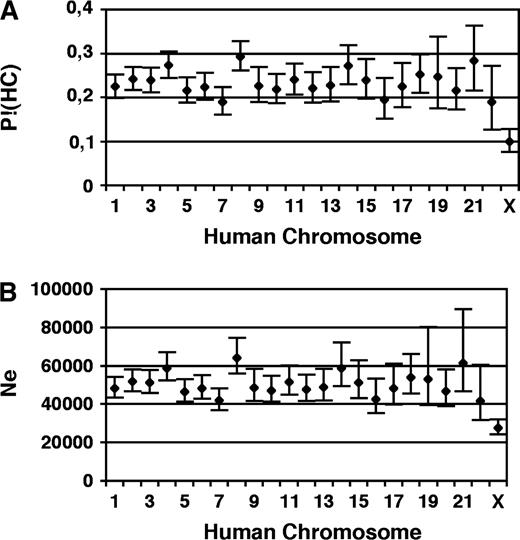
(A) Variation of the probability to observe a sequence tree significantly rejecting the human–chimp sister group (P!(HC)) among the human chromosomes. (B) Variation of the effective population size estimate of the human–chimp ancestral species among the human chromosomes. Bars denote the 95% CI.
The Paleodemographic History of Humans and Chimpanzees
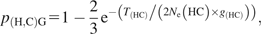
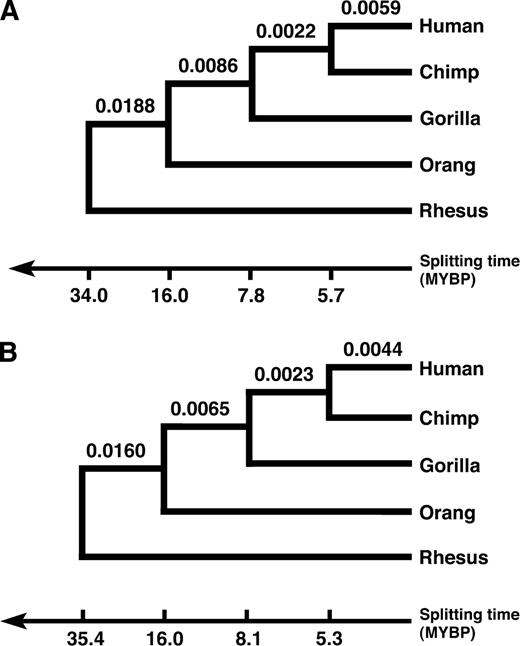
Sequence tree of the 5 species under study reconstructed from the autosomal sequences (A) and X chromosomal sequences (B), respectively. Splitting times were dated assuming the split of the orangutan lineage 16 MYBP. Trees are not drawn to scale.
The Evolutionary Age of Human Genetic Lineages
Subsequently, we identified those human genes in our data set for which exonic sequence overlapped with regions with an incongruent genealogy. A total of 125 genes were identified this way, 63 overlapped with regions placing chimpanzees closer to gorillas and 62 with regions supporting a human–gorilla grouping. A detailed list is given in the supplementary table 1 (Supplementary Material online).
Discussion
The evolutionary history of humans and the genetic relationships to their next closest relatives, the great apes, have been central to numerous studies in the past. Still, the picture about how and when humans emerged as a species from the common ancestor shared with chimpanzees remains fragmentary. In the present study, we have reanalyzed a data set of 23,210 alignments of human, chimpanzee, gorilla, orangutan, and rhesus DNA sequences from randomly chosen regions of the human genome (Patterson et al. 2006) using maximum likelihood. We infer that for about 23% of our data set chimpanzees are not the closest genetic relatives to humans. This figure is substantially smaller than previous estimates in the range of 40% based on far smaller data sets and where the varying extents of phylogenetic information in the data was not taken into account (Satta et al. 2000; Chen and Li 2001). However, it lies well in the range of the 18–29% as suggested by Patterson et al. (2006), who used a maximum parsimony approach to analyze the data. Nonetheless, one simplifying assumption in our analysis needs to be taken into account, which could bias the outcome of our analysis toward a slight underestimation of the fraction of incongruent trees. Given the limited length of the alignments available and the resulting average low number of phylogenetic informative positions, we had to assume that DNA sequences in the individual alignments have evolved according to a single sequence tree. Thus, recombination, which facilitates the alternation of varying phylogenies along a DNA sequence, had to be ignored. Evidence exists that regions in the human genome with an incongruent genealogy might be short compared with our average alignment length (630 bp) (Hobolth et al. 2007). In such a case, the phylogeny is likely to change within an alignment that contains incongruent sites. As a consequence, the alignment has an increased probability to be phylogenetically uninformative in our analysis. To assess the extent of underestimation, we analyzed the fraction of parsimoniously phylogenetic informative sites with respect to the human–chimp–gorilla subtree in our entire data set. A total of 23,510 sites support the grouping of humans and chimpanzees, whereas 9,596 sites (29%) suggest an incongruent genealogy. Because this approach assumes that every site evolves independently, and neglects homoplasy, which mimics an incongruent generalogy, 29% comprises the upper boundary of incongruent sites in our data set.
A Map of Our Genetic Ancestry
In view of the random character of the sampling strategy, our results indicate that roughly one-quarter of our genome shares no immediate ancestry with chimpanzees. To get an impression about the spatial distribution of such regions, we mapped the resolved sequence trees onto the human chromosomes (fig. 5 and supplementary fig. 2 [Supplementary Material online]). Incongruent sequence trees are present on all autosomes as well as on the X chromosome and display no general tendency for a regional clustering. Thus, these chromosomes emerge by and large as random assemblies of regions owning a distinct evolutionary relationship to the great apes. Reshuffling of parental chromosomal loci during meiosis has presumably acted to decouple the evolutionary histories of genetic regions located on the same DNA molecule (Paabo 2003; Hobolth et al. 2007). However, the probability to observe an incongruent sequence tree seems not to be the same throughout our genome. When we asses the fraction of incongruent sequence trees for the individual human chromosomes, values range between 18% and 29% with a mean of 23.6% for the human autosomes, and it is as low as 10% for the human X chromosomes (cf., fig. 3A). For the autosomes, we can only speculate about the causes leading to the observed variation in the fraction of incongruent trees. Likely explanations fall into 2 categories. The first one assumes that all autosomes share the same fraction of sites with an incongruent genealogy. However, the power to detect these sites differs significantly for each chromosome. As one possible scenario, we could imagine that recombination patterns may have varied among chromosomes in the human–chimpanzee ancestor. As a consequence, sites following an incongruent genealogy might be more scattered and thus more difficult to detect in our analysis, for one chromosome than for another. However, we find no correlation between the fraction of incongruent sequence trees and the fraction of unresolved trees for the individual autosomes. The second category of explanations assumes that the individual autosomes differ significantly in their fraction of sites with an incongruent genealogy. Such a deviation from the neutral model detailed in figure 1 could point toward the effect of selection. For example, regions under balancing selection (Charlesworth 2006) in the common ancestor of humans and chimpanzees have a higher probability to retain ancestral polymorphisms and, thus, would show up as regions in the human genome with an increased fraction of incongruent sequence trees. In turn, selective sweeps (Voight et al. 2006) in the human–chimp ancestral species would remove any ancestral polymorphisms and, thus, result in islands in our genome, where incongruent sequence trees are depleted. Notably, in figure 5 and supplementary figure 2 (Supplementary Material online) individual genomic regions can be seen that are conspicuously free of incongruent sequence trees.
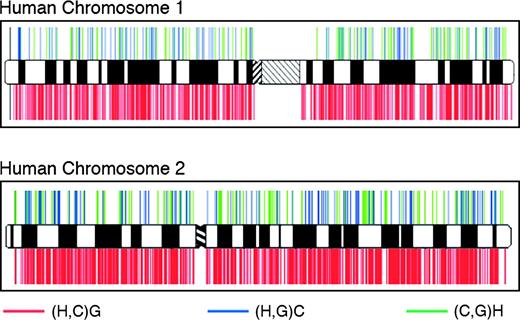
Distribution of resolved sequence trees recovering the monophyly of humans and the great apes along human chromosomes 1 and 2.
In summary, whatever causes the observed variation in the fraction of incongruent sequence trees, we can think of no influence resulting in an overestimation of this fraction. From this perspective, our figure of 23.6% incongruencies serves as a conservative baseline for the entire human autosomes. Any region deviating significantly from this figure is indicative of having an evolutionary history that differs from the genome average.
The situation is, on a first sight, different for the human X chromosome. So far, we have not taken into account its lower effective population size, which is only three-quarter that of the autosomes. Thus, a significant lower observation frequency of incongruent sequence trees on the X chromosome to the autosomes is not surprising.
Population Size of the Species Ancestral to Humans and Chimpanzees
From the observed frequency of incongruent sequence trees on the autosomes, we conclude that the species from which humans and chimpanzees emerged had a population size in the range of 49,000. This value appears in agreement with previous studies that chose different approaches to this question (Wall 2003; Hobolth et al. 2007). The estimate, however, essentially depends on the choice of the individual species’ generation times and the calibration of the molecular clock. For example, Hobolth et al. (2007) used a mean generation time as high as 25 years for all extant and ancestral species, a value that can be safely considered unrealistic. This choice essentially implies that their data support a substantially higher effective population size of the predecessor species of humans and chimpanzees than their stated figure of 50,000. Our assumption of g(HC) = 20 years can still be considered as too high and might reflect more the generation time of contemporary humans rather than that of the ancestral species we shared with chimps. Reducing g(HC) to 15 years increases e(HC) to 67,000 (95% CI 65,000–70,000). On the other hand, our dating for the split of the orangutan lineage (16 MYBP) used for calibrating the molecular clock might be too far back in time. A recent study on estimating the time points when the individual primate lineages have emerged places the split of the orangutan lineage 13 MYBP (Glazko and Nei 2003). Based on this splitting time, (HC) reduces to 1.7 Myr. With 20 years as an estimate for g(HC), e(HC), equals 41,000 (95% CI 39,000–42,000). Taking into account both our ignorance of g(HC) and T(HC), we conclude that the effective population size of the species ancestral to humans and chimpanzees was 49,000 with a range from 39,000 to 69,000. A comparison of these estimates with the effective population sizes assessed for the contemporary human and great ape populations (Kaessmann et al. 2001; Yu et al. 2001) reveals that considerably little has changed in the demographic history of chimpanzees and gorillas. In contrast, about a 5-fold reduced effective population size is observed for extant humans and bonobos. This adds a further line of evidence that humans—as must have bonobos—experienced a severe demographic bottleneck in their recent evolutionary history (Kaessmann et al. 2001).
The Peculiar Evolutionary History of the Human X Chromosome
The effective population size of the species ancestral to humans and chimpanzees estimated from the fraction of incongruent sequence trees observed on the X chromosome is 28,000 (95% CI 24,000–32,000). This value is significantly smaller than the expected Ne,X(HC)=3/4 × 49,000 = 36,750, based on the analysis of the autosomal data. Accordingly, the reduction in the observation frequency of incongruent sequence trees on the X chromosome compared with the autosomes cannot be explained by its lower effective population size alone (binomial test: P < 0.0001). Moreover, when we assess the splitting times from the concatenated X chromosomal alignments, these results also differ from those obtained from the autosomal data (fig. 4B). The split of humans and chimpanzees is dated more recent (5.4 MYBP), and the time the common ancestor of humans and chimpanzees has existed is about 700,000 years longer than the corresponding estimate from the autosomes. When we use the dating from the X chromosomal sequences to assess Ne,X(HC) from the fraction of incongruent sequence trees on the X chromosome, we estimated an ancestral population size of 37,000 (95% CI 32,000–43,000). This value is almost exactly the expected three-quarters of the population size estimated from the autosomes. Thus, the prolonged time span for the human–chimpanzee ancestor for the X chromosome suffices to explain its reduced fraction of incongruent sequence trees. To date, we can only speculate about the underlying cause of the marked difference in the evolutionary history of the autosomes and the X chromosomes. Rehybridization of human and chimpanzees subsequent to their initial separation into 2 different species, as suggested in Patterson et al. (2006) and Osada and Wu (2005), might serve as one explanation. Alternatively, recurrent selective sweeps in the human–chimp ancestral population occurring more frequently on the X than on the autosomes—due to the direct exposition of recessive mutations to selection in males—could result in a similar figure. The removal of ancestral polymorphisms by selective sweeps has 2 effects. First, it reduces the fraction of incongruent sequence trees. Second, it reduces the coalescent time for human and chimpanzee genetic lineages. By that it places the human–chimp split inferred from their genetic distances more recent, and thus, it prolongs the estimated time span the ancestral species to humans and chimpanzees has existed. However, alternative explanations might exist. In this context, the distribution of incongruent sequence trees on the X chromosome might be informative. If repeatedly selective sweeps acted on the X chromosome, we would expect extended regions on the X that are depleted of incongruent sequence trees. Presumably, no such clustering is expected when hybridization between humans and chimpanzees would have caused the observed scenario. Unfortunately, the number of 50 incongruent sequence trees we could map to the X chromosome is yet too small to allow for this analysis.
The Evolutionary Age of the Human Genome
Twenty-three percent of our genome sharing no immediate ancestry with chimpanzees has a further interesting implication. Necessarily, the corresponding genetic lineages must have split from their MRCA shared with any other species already prior to the speciation of the gorilla. Thus, a substantial part of the human genome began to evolve—in a today's point of view—“human specific” way long before humans emerged as a species. Obviously, in our analysis, we observe only 2/3 of such “old“ genetic lineages (fig. 1, dashed graphs) because the sequence trees of the remaining 1/3 agree with the species tree. Taking this into account, our results suggest that the ancestry of as much as 35% of our genome dates back to the ancient species we shared with the gorilla. What is the relevance for the evolution of functional sequences in the human genome? We observe incongruent sequence trees with frequencies around the genome-wide average in regions covered by genes and exons (cf., table 3 and supplementary table 1 [Supplementary Material online]). Furthermore, no overrepresentation of certain GO terms in the list of genes for which an incongruent sequence tree overlapped with an exon was observed (data not shown). Jointly, this suggests that the evolutionary relationships of human and chimpanzee sequences are by and large independent from the presence and function of genes and exons. From this figure, we conclude that roughly 1 out of 3 genes (35%) contain at least parts, which evolved human specific already in the progenitor species of humans, chimpanzees, and gorillas. The consequences are intriguing.
Despite extensive studies on early human evolution, it is still unclear when in our evolutionary history we split from the ancestral species shared with the chimpanzees. Particularly puzzling in this context is the apparent discrepancy between the dating of this split based on genetic evidences and the age of fossils, which have been—due to their display of certain human specific characteristics—assigned to a hominid species, that is a species more closely related to humans than to chimpanzees. The extent of genetic differences between humans and chimpanzees usually places the split of humans and chimpanzees around 5–6 MYBP (Chen and Li 2001; Glazko and Nei 2003; Kuroki et al. 2006), with a variation of at least 4 Myr across the genome (Barton 2006; Patterson et al. 2006). However, the current interpretation of the fossil record argues for the presence of hominids already 5.8 MYBP (Orrorin tugenensis, Senut et al. 2001, and Ardipithecus kadabba, WoldeGabriel et al. 2001) and presumably as early as 6.5–7.4 MYBP (Sahelanthropus tchadensis, Brunet et al. 2005). Hitherto, only a single attempt has been made to reconcile this discrepancy. Patterson et al. (2006) proposed a circuitous evolutionary scenario, where humans and chimpanzees separated initially prior to the emergence of S. tchadensis, explaining both the fossil record and the evolutionary older parts of the human genome. A later gene flow between the 2 species facilitated by rehybridization with the chimpanzee was then proposed to explain the evolutionary younger fraction of our genome. Our results, however, provide a far more parsimonious explanation.
The varying evolutionary ages of the human genome are to a large extent simply a consequence of the stochastic nature of the coalescent process determining the genealogy of human and chimpanzee genetic lineages (Barton 2006). More importantly, in view of the age of certain human genetic lineages (cf., supplementary table 1, Supplementary Material online), it seems mandatory to consider that a number of phenotypic characteristics nowadays judged as human-specific inventions (apomorphies) existed de facto already in the ancestral species of humans and chimpanzees. It is only because the corresponding genetic lineages were lost in our next relatives that these characters became confined to humans. The unequivocal assignment of fossil remains to a species more closely related to humans than to chimpanzees based on the presence of certain human-specific apomorphies should, therefore, be taken with a grain of salt. A similar point can be made in context of the recent discussion concerning the position of Australopithecus afarensis in the hominid phylogeny (Rak et al. 2007). This species has been proposed as the common ancestor of later hominines (Johanson and White 1979; White et al. 2006) including the genus Homo. However, the observation of an evolutionary derived ramal morphology of A. afarensis, resembling the status in gorilla, whereas contemporary humans and chimpanzee display the ancestral state, was taken as evidence to exclude A. afarensis from human ancestry (Rak et al. 2007).
The problem in using apomorphies for the reconstruction of phylogenetic relationships, however, extends beyond the classification of fossils. A nonnegligible fraction of human genes is expected to share an immediate ancestry with the gorilla or with the common ancestor of chimpanzees and gorillas. Because gene products essentially define the phenotype, we can expect a certain proportion of derived morphological characters to support the sister grouping of humans and gorillas, or chimpanzees and gorillas. This expectation is corroborated by comparative morphological studies between humans, chimpanzees, and gorillas (Shoshani et al. 1996; Collard and Wood 2000). For a number of phenotypic characters, either humans or chimpanzees share the derived character state with other great apes although the ancestral character state is still seen in the respective other species (supplementary table 2, Supplementary Material online). To water down the apparently contradictory character of such data, taxonomists proposing the human–chimp sister group status employed additional evolutionary scenarios, for example, the same derived character state might have arisen twice independently during evolution (Pilbeam 1986; Lockwood et al. 2004). Opponents of the human–chimp grouping, on the other hand, exploited such observations to promote alternative phylogenies of humans and the great apes (Schwartz 1984). However, our genome-wide comparison of DNA sequences between human and great ape species provides an alternative explanation, which easily resolves the discrepancy between the various schools favoring one over the other phylogeny. The random sorting of ancestral genetic polymorphisms that have a phenotypic polymorphism associated can explain why synapomorphies can be shared among species that are not each other's closest relatives.
In summary, our study highlights the extent and implications of the intertwined genetic relationships between humans, chimpanzees, and gorillas. Clearly, a comprehensive understanding of how humans evolved their unique characteristics, which distinguishes them from all other extant species, depends essentially on our knowledge of the evolutionary history of our genes. From this perspective, an extensive sequencing of the gorilla genome will be required to make full use of the chimpanzee genome sequence on the way toward a map of our genetic ancestry.
The authors wish to thank Heiko A. Schmidt and Vinh Le Sy for help in the Maximum Likelihood Analyses and Tanja Gesell and Steffen Klaere for helpful comments on the manuscript. The work was in part supported by the German National Genome Network grant 01GR0105. Financial support from the Wiener Wissenschafts- und Technologie-Fond is also acknowledged.
References
Author notes
Associate Editor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}