





Abstract
We implement a Bayesian Markov chain Monte Carlo algorithm for estimating species divergence times that uses heterogeneous data from multiple gene loci and accommodates multiple fossil calibration nodes. A birth-death process with species sampling is used to specify a prior for divergence times, which allows easy assessment of the effects of that prior on posterior time estimates. We propose a new approach for specifying calibration points on the phylogeny, which allows the use of arbitrary and flexible statistical distributions to describe uncertainties in fossil dates. In particular, we use soft bounds, so that the probability that the true divergence time is outside the bounds is small but nonzero. A strict molecular clock is assumed in the current implementation, although this assumption may be relaxed. We apply our new algorithm to two data sets concerning divergences of several primate species, to examine the effects of the substitution model and of the prior for divergence times on Bayesian time estimation. We also conduct computer simulation to examine the differences between soft and hard bounds. We demonstrate that divergence time estimation is intrinsically hampered by uncertainties in fossil calibrations, and the error in Bayesian time estimates will not go to zero with increased amounts of sequence data. Our analyses of both real and simulated data demonstrate potentially large differences between divergence time estimates obtained using soft versus hard bounds and a general superiority of soft bounds. Our main findings are as follows. (1) When the fossils are consistent with each other and with the molecular data, and the posterior time estimates are well within the prior bounds, soft and hard bounds produce similar results. (2) When the fossils are in conflict with each other or with the molecules, soft and hard bounds behave very differently; soft bounds allow sequence data to correct poor calibrations, while poor hard bounds are impossible to overcome by any amount of data. (3) Soft bounds eliminate the need for “safe” but unrealistically high upper bounds, which may bias posterior time estimates. (4) Soft bounds allow more reliable assessment of estimation errors, while hard bounds generate misleadingly high precisions when fossils and molecules are in conflict.
Introduction
The molecular clock assumption, that is, a constant evolutionary rate over time (Zuckerkandl and Pauling 1965), provides a simple yet powerful way of dating evolutionary events. Under the clock, the expected distance between sequences sampled from a pair of species (in units of expected numbers of substitutions) increases linearly with their time of divergence. When the clock is calibrated using external information about the geological ages of one or more nodes on the phylogeny (typically based on the fossil record), branch lengths estimated from sequences can be converted into geological times (Sanderson 1997; Rambaut and Bromham 1998; Yoder and Yang 2000; Ho et al. 2005). Early applications of the molecular clock to date species divergences typically use one calibration point, treated as known without error (Graur and Martin 2004; Hedges and Kumar 2004). However, fossil date estimates are not perfect and usually provide only an indication of the probability that species arose in some interval of time. Previous attempts to model this uncertainty assume the calibration age is uniformly distributed between two bounds—the probability that the date falls outside the interval is then zero (Thorne, Kishino, and Painter 1998).
“Hard” bounds, such as those imposed by a uniform prior, often overestimate the confidence in the fossil records. In particular, fossils often provide good lower bounds (i.e., minimum node ages), but not good upper bounds (maximum node ages). As a result, the researcher may be forced to use an unrealistically high upper bound to avoid precluding an unlikely (but not impossible) ancient age for the clade. This strategy is problematic as the bounds imposed in the prior may influence the posterior time estimation. Furthermore, a uniform distribution is unlikely to capture all the information about the likely age of a speciation event. For these reasons, more flexible distributions (with a mode, e.g.) and “soft” bounds (with nonzero probabilities everywhere) appear preferable. Finally, it is of interest to combine prior distributions from fossils with models of cladogenesis to allow a more complete description of the speciation process. This also allows one to examine the influence of the prior on divergence time estimates (e.g., the “robustness” of the inferences to the prior) by modifying parameters of the prior and examining the effect on the posterior.
Here, we present a new approach for incorporating fossil calibration information in the prior for divergence times for use in Bayesian estimation of divergence times. A range of flexible priors on fossil ages are combined with a birth-death process with species sampling to allow fossil information from multiple calibration points to be taken into account jointly when divergence times are inferred. We analyze real and simulated data sets to evaluate the performance of the new methods, especially in comparison with previous approaches that use hard bounds.
Theory
The Bayesian Framework
The topology of the rooted tree relating s species is assumed known and fixed. Aligned sequences are available at multiple loci, with the possibility that some species are missing at some loci. Our combined analysis accommodates differences in the evolutionary dynamics of different genes, such as different rates, different transition/transversion rate ratios, or different levels of rate variation among sites (Yang 2004). We assume that the divergence times are shared among different loci. We envisage that the methods will be applied to species data, so that recombination and lineage sorting are unimportant, and one set of divergent times applies to all loci. We assume the molecular clock, so that one rate r applies to all branches of the tree, although different loci can have different rates. Variable rates among sites within each locus are accommodated in the substitution model (Yang 1994). Here we illustrate the theory using one locus with one r. Let D be the sequence data, t be the s − 1 divergence times, and θ be the parameters in the substitution model and in the prior for divergence times t and rate r.
Prior Distribution of Divergence Times
Kishino, Thorne, and Bruno (2001) devised a recursive procedure for specifying f(t|θ), proceeding from ancestral to descendent nodes. A gamma density is used for the age of the root (t1 in the example tree of fig. 1), and a Dirichlet density is used to break the path from an ancestral node to the tip into time segments, corresponding to branches on that path. For example, along the path from the root to the bonobo (fig. 1), the five proportions (t1 − t2)/t1, (t2 − t3)/t1, (t3 − t4)/t1, (t4 − t5)/t1, and t5/t1 follow a Dirichlet distribution with equal means. Next, the two proportions (t2 − t6)/t2 and t6/t2 follow a Dirichlet distribution with equal means. Lower and upper bounds for ages of fossil calibration nodes, such as t2 and t4, are implemented by rejecting proposals that contradict such bounds. This is equivalent to specifying a uniform distribution for ages at calibration nodes. Using this strategy, Kishino, Thorne, and Bruno (2001) were able to calculate the prior ratio f(t*|θ)/f(t|θ) analytically, although not the prior density f(t|θ) itself.
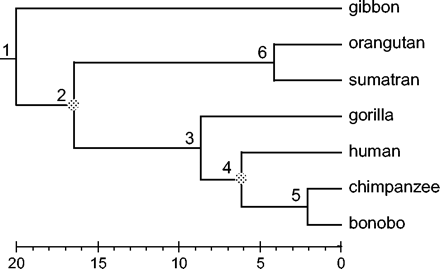
Phylogenetic tree for seven ape species used to explain priors for divergence times in the Bayesian methods. This tree is also used to analyze the mitochondrial data set of Cao et al. (1998), with nodes 2 and 4 used as fossil calibrations. The branches are drawn to show posterior means of divergence times estimated in the Bayesian analysis (table 3, “All, HKY + G”). Estimated times are in millions of years before present. The HKY + G model was assumed to analyze the three codon positions simultaneously, accounting for their differences.
It is difficult to implement flexible priors for fossil calibration ages using this approach as even the prior ratio does not then appear analytically tractable. To implement soft bounds or otherwise flexible priors for fossil calibrations, we use instead the birth-death process (Kendall 1948) generalized to account for species sampling (Rannala and Yang 1996; Yang and Rannala 1997). Previous use of the same model in Bayesian time estimation (Aris-Brosou and Yang 2002, 2003) considered only one fossil calibration and one gene locus. It may be noted that the birth-death process is similar to the coalescent process widely used in population genetics. However, the latter specifies trees with very long internal branches, which may be unrealistic for species phylogenies.
Prior Densities for Fossil Calibration Times
Constraints on the ages of nodes from fossil or geological data are incorporated in the analysis through the prior f(tC|C). The separation of the calibration information f(tC|C) from the birth-death process prior in equation (12) enables us to specify arbitrarily flexible constraints. We note that use of fossils to specify calibration information for molecular clock dating is a complicated process. First, determining the date of a fossil is prone to errors, such as experimental errors in radiometric dating or assignment of the fossil to the wrong stratum. Second, placing the fossil correctly on the phylogeny can be very complex. For example, a fossil may be clearly ancestral to a clade, but by how much the age of the fossil species precedes the age of the common ancestor of the clade may be hard to determine. Misinterpretations of character state changes may also cause a fossil to be assigned to a wrong lineage. For example, a fossil presumed to be ancestral may in fact represent an extinct side branch and is not directly relevant to the age of the clade concerned. We make no attempt to deal with such complexities here. Instead, we describe a method that enables the researcher to incorporate any statistical distribution to describe uncertainties in the age of a calibration node and leave it to the individual to choose an appropriate prior for the problem at hand.
Most often, calibration information is in the form of lower and upper bounds. The problem with hard bounds is that any age outside the prior bounds will have posterior probability zero, whatever the data. We thus prefer soft bounds that allow small but positive probabilities outside the bounds. We have implemented four kinds of constraints, as follows.
- Lower bound (t > tL). We let the density decline toward zero from t = tL according to a power distribution, with a total probability 2.5%.We use θ = log(0.1)/log(0.9) = 21.85 to achieve a relatively sharp decay (fig. 2a); this means that 90% of the density left of tL is within 10% of tL (i.e., between 0.9tL and tL). For the region t ≥ tL, we use an improper uniform prior. We suggest that one should not use lower bounds alone in the analysis. An example lower bound is shown in figure 2a. The node age is at least 12 Myr, represented by “>0.12,” with one time unit to be 100 Myr.(15)\[f(t){=}\left\{\begin{array}{ll}0.025{\times}\frac{\mathrm{{\theta}}}{t_{L}}\left(\frac{t}{t_{L}}\right)^{\mathrm{{\theta}}{-}1},&\mathrm{if}{\,}t{<}t_{L},\\0.025{\times}\frac{\mathrm{{\theta}}}{t_{L}}&\mathrm{if}{\,}t{\geq}t_{L}.\end{array}\right.\]
- Upper bound (t < tU). We use a uniform distribution in the interval (0, tU) with 97.5% of probability density and an exponential decay for t > tU with probability density 2.5%.We fix θ = 0.975/(0.025tU) so that f(t) is continuous at t = tU. Figure 2b shows the density for the upper bound t < 16 Myr, represented as “<0.16.”(16)\[f(t){=}\left\{\begin{array}{ll}\frac{0.975}{t_{U}},&\mathrm{if}{\,}t{<}t_{U},\\0.025\mathrm{{\theta}}{\,}\mathrm{e}^{{-}\mathrm{{\theta}}(t{-}t_{U})},&\mathrm{if}{\,}t{\geq}t_{U}.\end{array}\right.\]
- Lower and upper bounds (tL < t ≤ tU). We use a uniform distribution in the region tL < t ≤ tU with 95% probability. On the left side (t < tL), we have a power decay, while on the right side (t > tU), we have an exponential decay.We fix θ = 0.95tL/(0.025(tU−tL)) and θ2 = 0.95/(0.025(tU−tL)) so that f(t) is continuous at tL and tU. Figure 2c shows the density for the bounds 12 Myr < t < 16 Myr, represented as “>0.12 < 0.16.”(17)\[f(t){=}\left\{\begin{array}{ll}0.025{\times}\frac{\mathrm{{\theta}}_{1}}{t_{L}}\left(\frac{t}{t_{L}}\right)^{\mathrm{{\theta}}_{1}{-}1},&\mathrm{if}{\,}0{<}t{\leq}t_{L}\\\frac{0.95}{t_{U}{-}t_{L}},&\mathrm{if}{\,}t_{L}{<}t{\leq}t_{U},\\0.025{\times}\mathrm{{\theta}}_{2}{\,}\mathrm{exp}{\{}{-}\mathrm{{\theta}}_{2}(t{-}t_{U}){\}},&\mathrm{if}{\,}t{>}t_{U}.\end{array}\right.\]
- Gamma distributed prior. If a most likely age t* is provided for a calibration node as well as lower and upper bounds tL and tU, we use a gamma distribution prior. The density isParameters α and β are calculated from tL, tU, and t*. We consider it important for the prior density to have a positive (nonzero) mode and thus fix the mode to the most likely age: (α − 1)/β = t* with α > 1. We then estimate α and β by matching as closely as possible tL and tU with the 2.5% and 97.5% percentiles of the gamma distribution. Note that the gamma distribution has a heavier right tail than left tail, although the distribution approaches the normal density when both α and β are large. A close match between the gamma and tL and tU can be achieved when the most likely value is close to the midpoint of the two bounds but slightly to the left. The gamma density of figure 2d is specified by “>0.12 = 0.139 < 0.16,” indicating that the node age is between tL = 12 Myr and tU = 16 Myr and is most likely around 14 Myr. The gamma distribution fitted is G(186.9, 1,337.7), which has the mean 0.14 and tail probabilities Pr(t < tL) = 2.24% and Pr(t > tU) = 2.76%.(18)\[f(t;\mathrm{{\alpha}},\mathrm{{\beta}}){=}\frac{\mathrm{{\beta}}^{\mathrm{{\alpha}}}{\,}\mathrm{e}^{{-}\mathrm{{\beta}}t}t^{\mathrm{{\alpha}}{-}1}}{{\Gamma}(\mathrm{{\alpha}})}.\]
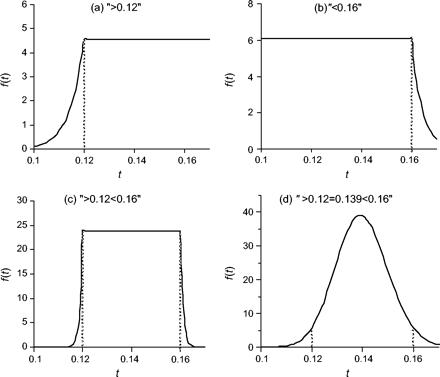
Probability densities implemented to describe uncertainties in fossil dates: (a) lower bound, specified as “>0.12”; (b) upper bound, specified as “<0.16” (c) lower and upper bounds, specified as “>0.12 < 0.16” and (d) gamma distribution G(186.9, 1337.7), specified as “>0.12 = 0.139 < 0.16.”
We note that in addition to the prior densities of times for the fossil calibration nodes, there is an intrinsic constraint on node ages; that is, the age of an ancestral node must be older than the age of a descendent node. Thus, the marginal prior density of the calibration node ages is not simply the product of the densities discussed above but is the joint density conditional on these intrinsic constraints. The difference between the true joint density and the product of the marginal densities can be large if the prior bounds for ancestral and descendent nodes overlap.
We stress that any sensible dating analysis should use at least one upper bound (maximum age) and at least one lower bound (minimum age) as fossil calibrations, although the bounds do not have to be on the same node. A single gamma distribution also achieves a similar effect as a lower and an upper bound. Uncertainties in most fossils appear to be best described by a highly asymmetrical distribution, with an extremely long right tail extending to earlier times, like our lower bounds. However, one should not conduct analysis using such calibrations only; an upper bound setting the maximum age of a node is essential.
Inference with Infinite Data
It is important to realize that for a specified set of fossil calibration bounds, progressively increasing the number of sites in the sequence will not reduce the errors in posterior estimates of the divergence times to zero. The sequence data provide information about the distances (or branch lengths) separating taxa but not the times and rates separately. Even if we have infinitely long sequences and can estimate branch lengths with no errors, uncertainties will remain in the posterior time estimates.
A Normal Distribution Example
We draw an analogy to a simple problem of estimating the means of two normal distributions. Suppose the data are y = {y1, y2, …, yn}, an independent and identically distributed sample of size n from N(μ, 1), with mean μ = μ1 + μ2. We are interested in the marginal posterior μ1|y. We assign priors μ1 ∼ N(−1, v1) and μ2 ∼ N(1, v2). Note that both in this example and in time estimation, the likelihood depends on a function of two parameters (the sum of two means in the normal example and the product of time and rate in time estimation) but not on the two parameters separately. Thus, both involve an identifiability problem. It can easily be shown (see Appendix B) that when n → ∞,
Asymptotic Posterior Distribution of Divergence Times
A few remarks on these results are in order. First, with infinitely many sites in the sequence, the posterior does not converge to a point mass on the true parameter values. Rather, the posterior converges to a one-dimensional distribution, signifying that the uncertainty still remains. In this limiting case, the branch lengths are estimated without error, and the enforcement of the molecular clock means that given the rate all divergence times are fully determined. Second, the posterior means for all node ages tj will lie on a straight line when plotted against the true ages, as will the percentiles and credibility intervals (CIs). In real data analysis, one can plot the CIs against the posterior means of divergence times to assess how well the results fit straight lines and whether the amount of sequence data is nearly saturated. Third, if only one fossil calibration is available, the posterior density for that node will approximately be the prior on the calibration, while the posterior for other divergence times will be determined though linear transformations of this prior. With more than one calibration, the posterior for the age of each calibration node will be more informative than the prior on that node because information is pooled across nodes. For a given sequence length n, the ratio of the CI widths wn/w∞, where wn is the CI width on the rate (or any node age tj) for n sites and w∞ is the asymptotic CI width when n → ∞, measures whether increasing sequence data further is likely to improve the precision of posterior time estimates. When the ratio is close to 1, the sequence data is nearly saturated.
Computer Simulation
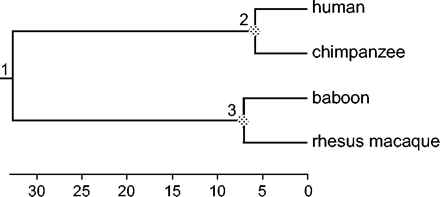
We conducted a computer simulation to examine the performance of soft bounds, in comparison with hard bounds. We implemented hard bounds by using soft bounds with a very small tail probability 10−299 instead of 0.025. Our interest was in the effect of increasing sequence length on the accuracy of divergence time estimates. Sequence data were generated using the EVOLVER program in the PAML package (Yang 1997) and the model tree of figure 3. The branch lengths conform to a molecular clock, with the distance from the root to the present time being one expected nucleotide substitution per site. The JC model (Jukes and Cantor 1969) was used in both simulation and analysis. We suppose the rate is 1 nt substitution per time unit, so that the true ages of nodes 1 (the root), 2, …, 8 are 1, 0.7, 0.2, 0.4, 0.1, 0.8, 0.3, and 0.05 (fig. 3). If one time unit is 100 Myr, then the age of the root is 100 Myr, and the substitution rate is 10−8 substitutions per site per year.
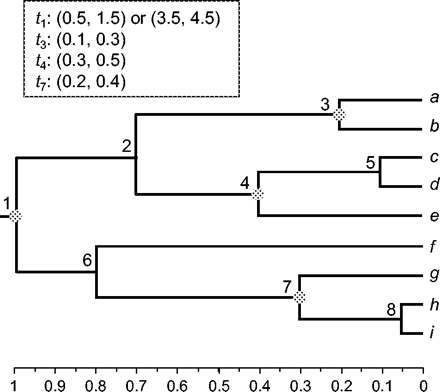
A tree of nine species used in computer simulation to examine the performance of soft and hard bounds. The tree conforms to the molecular clock, with the amount of sequence change from the root to the present time to be one substitution per site. For divergence time estimation, the rate is assumed to be one change per time unit, so that the true times for nodes 1, 2, …, 8 are 1, 0.7, 0.2, 0.4, 0.1, 0.8, 0.3, and 0.05. Nodes 1, 3, 4, and 7 are used as fossil calibrations, with good fossils always available for nodes 3, 4, and 7, but the root (node 1) has either a good fossil (with bounds 0.5, 1.5) or a bad fossil (with bounds 3.5, 4.5).
For Bayesian divergence time estimation, nodes 1, 2, 4, and 7 are used as fossil calibration nodes (fig. 3). It is assumed that good fossils are always available for nodes 3, 4, and 7, specified using the bounds (0.1, 0.3) for t3, (0.3, 0.5) for t4, and (0.2, 0.4) for t7. The root (node 1) has either a good fossil or a bad fossil, with the bounds (0.5, 1.5) for the good fossil or (3.5, 4.5) for the bad fossil; note that the true age is t1 = 1. The prior for divergence times is specified using the birth-death process with species sampling, with the birth and death rates λ = μ = 2, and sampling fraction ρ = 0.1. The kernel density for those parameters (eq. 4) is nearly flat between t1 = 0 and 1, suggesting that the node ages t−1 = t2, t3, …, t8 are ordered random variables from a nearly uniform distribution (see fig. 2 of Yang and Rannala [1997] for plots of such densities). The substitution rate is assigned a gamma prior G(2, 2), with mean 1 and variance ½.
Estimation with Good Fossil Calibrations
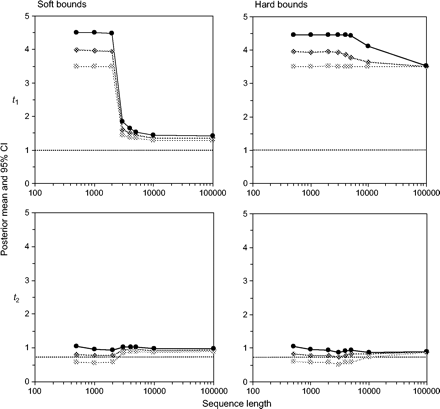
Posterior means and 95% CIs for divergence times t1 and t2 (see fig. 3) are plotted against the sequence length n in figure 4 for the good fossils. Results for all divergence times t = t1, t2, …, t8 are presented for a large data set with 106 sites in figure 5. First, we consider the performance of soft and hard bounds when only good fossil calibrations are used. Figure 4 shows that there was essentially no difference between soft and hard bounds. For both, the true times were close to the posterior means and well within the 95% CIs. With n = 106 sites, the posterior means for the times t lay on a straight line, so did the 2.5% percentiles and the 97.5% percentiles (fig. 5). For soft bounds, the posterior mean of t1 was 1.05, with the 95% CI to be (0.77, 1.26) (fig. 5a), which were very close to the theoretical limits of 1.06 (0.78, 1.25) when n → ∞ (eq. 25). The corresponding results for hard bounds were 1.05 (0.77, 1.24) for n = 106 sites (fig. 5b) and 1.06 (0.78, 1.24) for n → ∞. The soft and hard bounds produced nearly identical results. Note that with no data, the posterior CI for t1 is the prior interval, approximately (0.5, 1.5), with a width of 1. With the increase of the sequence length, the posterior CI became narrower. When n → ∞, the width of the 95% CI is 0.46, so that the interval is reduced by only one-half relative to the prior on the calibration age by using infinitely long sequences. Thus, at this limit, every 1 Myr of divergence time adds 0.46 Myr to the 95% CI. Indeed, the influence of increased sequence data was essentially saturated at n = 10,000 sites, when the 95% CI width was 0.49, very close to the width 0.46 at n → ∞.
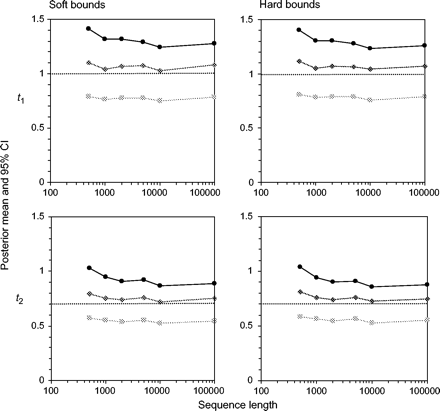
Posterior means and 95% CIs of divergence times t1 and t2 (fig. 3) plotted against the sequence length when all fossils are good. The three points at each sequence length are, from bottom to top, the 2.5% percentile, the mean, and the 97.5% percentile of the posterior distribution. Fossil calibrations are shown in figure 3, with the bound for t1 to be (0.5, 1.5). The true times are t1 = 1 and t2 = 0.7, indicated by the dotted lines. The results for all times t1–t8 when sequence length is n = 106 are shown in figure 5a and b.
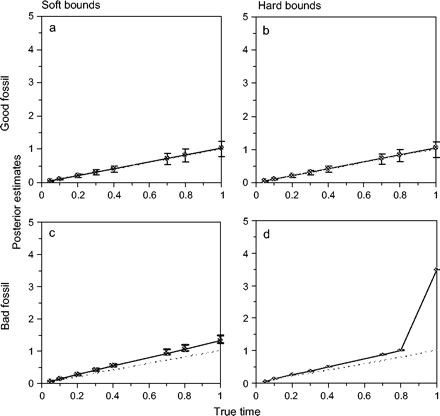
Posterior mean and 95% CI plotted against the eight true divergence times in a simulated data set with n = 106 sites. See legends to figures 3 and 4 for simulation details. (a) and (b) are for “good fossil” (see fig. 4), and (c) and (d) are for “bad fossil” (see fig. 6). Also (a) and (c) are for soft bounds and (b) and (d) are for hard bounds.
Estimation with a Bad Fossil Calibration
Divergence time estimates obtained using the bad fossil calibration prior are shown in figure 6. The root age t1 was specified to be within the bounds (3.5, 4.5), while the true age is 1; that is, if the true age is 100 Myr, the grossly misspecified fossil bounds are from 350 to 450 Myr! In small data sets with n ≤ 2,000 sites, the soft and hard bounds produced similar results. The posterior 95% CI for the root age was close to the prior interval (3.5, 4.5), while the posterior for other divergence times, such as t2 (fig. 6), was closer to the true times due to the influence of the three good fossils at nodes 3, 4, and 7. However in large data sets with n ≥ 3,000 sites, soft and hard bounds behaved very differently. With soft bounds, the sequence data and the good prior calibration intervals appeared to overcome the poor calibration interval at the root so that posterior estimates of t1 improved considerably (fig. 6). At n = 106, the posterior mean for t1 was 1.33, with the 95% CI to be (1.25, 1.41). The theoretical limits at n → ∞ were 1.33 (1.28, 1.37). Thus, with infinite sites, all divergence times t would be overestimated by 33%, with the 95% CI width to be 0.09 Myr for every 1 Myr of true divergence. The true times were well outside the CIs.
Posterior means and 95% CIs of divergence times t1 and t2 (fig. 3) plotted against the sequence length when one bad fossil is used. Fossil calibrations are shown in figure 3, with the bounds for t1 to be (3.5, 4.5), representing a bad fossil. See legend to figure 4 for more details. The results for all times t1–t8 when sequence length is n = 106 are shown in figure 5c and d.
When hard bounds were used, the sequence data and the fossil calibrations were in direct conflict. With the sequence length n > 105, the posterior mean of t1 converged to the lower bound (3.5), while the 95% CI became virtually a point (figs. 5d and 6). The posterior mean for t1 is grossly wrong, and the high precision is misleading. Posterior means of other divergence times were not seriously wrong due to the influence of the good fossils. However, their posterior CIs still converged to single points, with misleadingly high precisions. Thus, the extremely narrow CIs, which are at the prior bounds, reflect conflicts among fossil calibrations or between fossils and the molecular data rather than high precision of estimation. Note that with hard bounds the sequence data and the fossils are in contradiction so that the limiting theory for n → ∞ cannot be applied. For example, the infinite data suggest that t1 = 5t3 (see fig. 3), so with the upper bound 0.3 for t3, it is impossible for t1 to be older than 1.5. Similarly, consideration of upper bounds at t4 and t7 suggest that t1 should not be older than 1.25 or 1.33. Thus, the specified lower bound t1 > 3.5 causes contradictions among the fossils. It is apparent that systematic errors in fossil calibrations will deflate posterior confidence intervals for divergence times when using prior calibration intervals with either soft or hard bounds, although the problem is much more severe with hard bounds. Residual uncertainties due to finite sequence length might mask such trends, however, and should also be examined.
Analysis of Primate Data Sets
We analyze two data sets to test the new algorithms incorporating soft bounds in fossil calibrations. The first consists of five genomic contigs from two primates and two Old World monkeys (Steiper, Young, and Sukarna 2004). We analyze the five loci both separately and as a combined data set. The second data set consists of the 12 protein-coding genes from the mitochondrial genome from seven ape species (Cao et al. 1998; Yang, Nielsen, and Hasegawa 1998). We merge the 12 genes into one supergene as they have similar evolutionary dynamics but accommodate the huge differences among the three codon positions. The nucleotide substitution model of Hasegawa, Kishino, and Yano (HKY) (1985) was used together with the discrete gamma model of rate variation among sites, with five rate categories being used (Yang 1994). The model is represented as HKY + G and accounts for unequal transition and transversion rates, unequal nucleotide frequencies, and unequal rates among sites. We assign the gamma prior G(6, 2) with mean 3 and variance 1.5 for the transition/transversion rate ratio κ and the gamma prior G(1, 1) for the gamma shape parameter α. The nucleotide frequencies are estimated using the observed frequencies.
In the combined analysis (of all five loci in data set 1 and of all three codon positions in data set 2), the model assumes different rates and different substitution parameters (κ, α, and base frequencies) among site partitions (Yang 1996). The JC model (Jukes and Cantor 1969) is used for comparison as well. The substitution rate for each site partition is assigned the gamma distribution G(2, 2). We used the same priors for both data sets and, as far as possible, for the computer simulation, to simplify the description. The data sets are very informative about substitution parameters such as κ, α, and rates, and these priors had very little effect on the posterior estimates. In the birth-death process with species sampling, we fix the birth and death rates at λ = μ = 2, with the sampling fraction ρ = 0.1, as in the computer simulation discussed above. However, we also used two other sets of values for λ, μ, and ρ to examine the effects of the prior for divergence times on posterior estimation. We implement in the computer program an option of assigning priors on λ, μ, and ρ and integrating out those parameters using a hierarchical Bayesian approach.
We conducted initial runs to fine-tune the step lengths for proposing changes in the Metropolis-Hastings algorithm and to determine how long the Markov chain has to be run to reach convergence. The results presented below were obtained by discarding 10,000 iterations as the burn-in, followed by 100,000 iterations, sampling every five iterations. Every analysis was conducted by running the chain at least twice, using different starting values, to confirm consistency between runs.
The Date Set of Steiper, Young, and Sukarna
We analyzed the data of Steiper, Young, and Sukarna (2004), which consist of five genomic contigs from four species: human (Homo sapiens), chimpanzee (Pan troglodytes), baboon (Papio anubis), and rhesus macaque (Macaca mulatta). The contigs (referred to as A, B, C, D, and E) range from ∼12 to 64 kbp long. See Steiper, Young, and Sukarna (2004) for the GenBank accession numbers. The phylogeny for the species is shown in figure 7. Steiper, Young, and Sukarna (2004) conducted likelihood ratio tests (LRTs) of the molecular clock for each of the five contigs. The loci that passed the test were then analyzed using the quartet dating approach of Rambaut and Bromham (1998), fixing the age of the human-chimpanzee divergence at either 6 or 7 Myr and the age of the baboon-macaque divergence at either 5 or 7 Myr. Thus, each analysis fails to accommodate uncertainties in fossil dates, but the range of estimates produced in several analyses fixing fossil node ages at different constants provides an intuitive assessment of the effect of fossil uncertainties.
The four-species tree for the data of Steiper, Young, and Sukarna (2004), with branches drawn in proportion to the posterior means of divergence times estimated from the data (table 2, “All combined”). Fossil calibrations at nodes 2 and 3 are available. See text for details.
LRTs of the Clock and Maximum Likelihood Estimation of Divergence Dates
We conducted a simple likelihood analysis to estimate model parameters reflecting basic properties of the evolutionary process and to obtain results for comparison with the Bayesian analysis. We apply two LRTs of the molecular clock and examined the corresponding maximum likelihood estimates (MLEs) of divergence times for each contig (table 1). We note that it is also possible to apply a Bayesian approach for testing the clock (Suchard, Weiss, and Sinsheimer 2003). The first test we conducted is the commonly used LRT of the clock described by Felsenstein (1981). The alternative no-clock model estimates five branch lengths on the unrooted tree, while the null-clock model estimates three node ages on the rooted tree—the distances from the three internal nodes to the present times. No fossil information is used in this test of the clock model. Twice the log likelihood difference is compared with a χ2 distribution with df = 2. This test examines whether the human and chimpanzee are equidistant from their common ancestor and whether the baboon and macaque are equidistant from their common ancestor (fig. 7). This test failed to reject the clock in any analysis under either JC or HKY + G (table 1). The second test of the clock uses the same alternative model, but the null model uses the two fossil calibrations to calculate the log likelihood, estimating the age of the root and the substitution rate. The test thus has three degrees of freedom. Steiper, Young, and Sukarna (2004) used this test, considering all combinations of the ages at the two calibration nodes 2 and 3 in the tree: (6, 5), (6, 7), (7, 5), (7, 7). If the fossil dates are correct, this test may be expected to be more powerful. If the fossil dates are incorrect, this test may mistake the unreliability of the fossil dates as violation of the molecular clock. We used the fossil dates (7, 6) to conduct the test (table 1). The clock was rejected in contigs B and D and in the combined analysis. In contig C, the clock was marginally rejected by this test.
LRT Statistic of the Molecular Clock and MLEs of Ages and Rate Under JC and HKY + G Models for the Data of Steiper, Young, and Sukarna
Data | Δℓ (test 1) | Δℓ (test 2) | Root Age | Rate | \(\mathrm{{\hat{{\kappa}}}}\) | \(\mathrm{{\hat{{\alpha}}}}\) |
|---|---|---|---|---|---|---|
| JC | ||||||
| A | 0.76 | 0.81 | 28.2 | 7.4 | ||
| B | 1.88 | 24.95** | 29.6 | 8.3 | ||
| C | 2.10 | 4.40* | 34.2 | 5.8 | ||
| D | 0.22 | 35.64** | 34.8 | 6.1 | ||
| E | 0.68 | 3.19 | 37.9 | 5.6 | ||
| All concatenated | 1.71 | 49.89** | 32.8 | 6.6 | ||
| All, combined | 1.71 | 49.89** | 32.8 | |||
| HKY + G | ||||||
| A | 0.83 | 0.87 | 28.9 | 7.5 | 4.50 | 0.82 |
| B | 1.60 | 24.57** | 30.4 | 8.4 | 4.09 | 1.24 |
| C | 2.17 | 4.47* | 34.6 | 5.8 | 3.50 | 2.83 |
| D | 0.14 | 35.43** | 36.1 | 6.2 | 4.77 | 0.62 |
| E | 0.49 | 2.93 | 39.3 | 5.6 | 4.29 | 0.72 |
| All concatenated | 1.31 | 49.20** | 33.8 | 6.6 | 4.41 | 0.79 |
| All combined | 1.30 | 49.17** | 33.8 | 6.7 |
Data | Δℓ (test 1) | Δℓ (test 2) | Root Age | Rate | \(\mathrm{{\hat{{\kappa}}}}\) | \(\mathrm{{\hat{{\alpha}}}}\) |
|---|---|---|---|---|---|---|
| JC | ||||||
| A | 0.76 | 0.81 | 28.2 | 7.4 | ||
| B | 1.88 | 24.95** | 29.6 | 8.3 | ||
| C | 2.10 | 4.40* | 34.2 | 5.8 | ||
| D | 0.22 | 35.64** | 34.8 | 6.1 | ||
| E | 0.68 | 3.19 | 37.9 | 5.6 | ||
| All concatenated | 1.71 | 49.89** | 32.8 | 6.6 | ||
| All, combined | 1.71 | 49.89** | 32.8 | |||
| HKY + G | ||||||
| A | 0.83 | 0.87 | 28.9 | 7.5 | 4.50 | 0.82 |
| B | 1.60 | 24.57** | 30.4 | 8.4 | 4.09 | 1.24 |
| C | 2.17 | 4.47* | 34.6 | 5.8 | 3.50 | 2.83 |
| D | 0.14 | 35.43** | 36.1 | 6.2 | 4.77 | 0.62 |
| E | 0.49 | 2.93 | 39.3 | 5.6 | 4.29 | 0.72 |
| All concatenated | 1.31 | 49.20** | 33.8 | 6.6 | 4.41 | 0.79 |
| All combined | 1.30 | 49.17** | 33.8 | 6.7 |
NOTE.—The null model in test 1 assumes that each tip in the tree is equidistant from the root (fig. 7). The null model in test 2 assumes the clock and also fits two fossil calibrations to the tree: 7 Myr for the human-chimpanzee divergence and 6 Myr for the baboon-macaque divergence. In both tests, the alternative is the no-clock model, with five branch lengths in the unrooted tree as parameters. Significance is indicated by * for P < 5% or ** for P < 1%. Rate is ×10−10 substitutions per site per year.
LRT Statistic of the Molecular Clock and MLEs of Ages and Rate Under JC and HKY + G Models for the Data of Steiper, Young, and Sukarna
Data | Δℓ (test 1) | Δℓ (test 2) | Root Age | Rate | \(\mathrm{{\hat{{\kappa}}}}\) | \(\mathrm{{\hat{{\alpha}}}}\) |
|---|---|---|---|---|---|---|
| JC | ||||||
| A | 0.76 | 0.81 | 28.2 | 7.4 | ||
| B | 1.88 | 24.95** | 29.6 | 8.3 | ||
| C | 2.10 | 4.40* | 34.2 | 5.8 | ||
| D | 0.22 | 35.64** | 34.8 | 6.1 | ||
| E | 0.68 | 3.19 | 37.9 | 5.6 | ||
| All concatenated | 1.71 | 49.89** | 32.8 | 6.6 | ||
| All, combined | 1.71 | 49.89** | 32.8 | |||
| HKY + G | ||||||
| A | 0.83 | 0.87 | 28.9 | 7.5 | 4.50 | 0.82 |
| B | 1.60 | 24.57** | 30.4 | 8.4 | 4.09 | 1.24 |
| C | 2.17 | 4.47* | 34.6 | 5.8 | 3.50 | 2.83 |
| D | 0.14 | 35.43** | 36.1 | 6.2 | 4.77 | 0.62 |
| E | 0.49 | 2.93 | 39.3 | 5.6 | 4.29 | 0.72 |
| All concatenated | 1.31 | 49.20** | 33.8 | 6.6 | 4.41 | 0.79 |
| All combined | 1.30 | 49.17** | 33.8 | 6.7 |
Data | Δℓ (test 1) | Δℓ (test 2) | Root Age | Rate | \(\mathrm{{\hat{{\kappa}}}}\) | \(\mathrm{{\hat{{\alpha}}}}\) |
|---|---|---|---|---|---|---|
| JC | ||||||
| A | 0.76 | 0.81 | 28.2 | 7.4 | ||
| B | 1.88 | 24.95** | 29.6 | 8.3 | ||
| C | 2.10 | 4.40* | 34.2 | 5.8 | ||
| D | 0.22 | 35.64** | 34.8 | 6.1 | ||
| E | 0.68 | 3.19 | 37.9 | 5.6 | ||
| All concatenated | 1.71 | 49.89** | 32.8 | 6.6 | ||
| All, combined | 1.71 | 49.89** | 32.8 | |||
| HKY + G | ||||||
| A | 0.83 | 0.87 | 28.9 | 7.5 | 4.50 | 0.82 |
| B | 1.60 | 24.57** | 30.4 | 8.4 | 4.09 | 1.24 |
| C | 2.17 | 4.47* | 34.6 | 5.8 | 3.50 | 2.83 |
| D | 0.14 | 35.43** | 36.1 | 6.2 | 4.77 | 0.62 |
| E | 0.49 | 2.93 | 39.3 | 5.6 | 4.29 | 0.72 |
| All concatenated | 1.31 | 49.20** | 33.8 | 6.6 | 4.41 | 0.79 |
| All combined | 1.30 | 49.17** | 33.8 | 6.7 |
NOTE.—The null model in test 1 assumes that each tip in the tree is equidistant from the root (fig. 7). The null model in test 2 assumes the clock and also fits two fossil calibrations to the tree: 7 Myr for the human-chimpanzee divergence and 6 Myr for the baboon-macaque divergence. In both tests, the alternative is the no-clock model, with five branch lengths in the unrooted tree as parameters. Significance is indicated by * for P < 5% or ** for P < 1%. Rate is ×10−10 substitutions per site per year.
Table 1 shows the MLEs of the age of the root obtained under the clock model, assuming that nodes 2 and 3 are 7 and 6 Myr old. Note that the likelihood analysis fails to accommodate uncertainties in the fossil calibrations. The estimates were similar to those obtained by Steiper, Young, and Sukarna (2004). There were no systematic differences in the time estimates between contigs that conform to the clock (A and E) and contigs that violate the clock (B, C, D). Thus, we use all five contigs in our Bayesian analysis below.
Bayesian Divergence Time Estimation
We then apply the Bayesian method described in this paper. We used the gamma distribution to specify the two fossil calibration dates. The age of the human-chimpanzee divergence was assumed to be between 6 and 8 Myr, with the most likely date to be 7 Myr (Brunet et al. 2002). We specified the prior as “>0.06 = 0.0693 < 0.08” and fitted the gamma distribution G(186.2, 2672.6), so that the prior mean was 7 Myr, and the tail probabilities were Pr(t2 < 6) = 2.5% and Pr(t3 > 8) = 2.5%. The second calibration is for the divergence of baboon and macaque. We assumed that the date is between 5 and 7 Myr, most likely at 6 Myr (Delson et al. 2000). This was specified as “>0.05 = 0.0591 < 0.07,” and the gamma prior fitted was G(136.2, 2286.9), with mean at 6 Myr and tail probabilities 2.6% and 2.4%. See Steiper, Young, and Sukarna (2004) and Raaum et al. (2005) for reviews of relevant fossil data.
The posterior means and 95% CIs for divergence times obtained from the separate and combined analyses are shown in table 2. The posterior means were virtually identical to the MLEs under the clock model (table 1) and similar to the MLEs obtained by Steiper, Young, and Sukarna (2004). However, the Bayesian analysis has the advantage of providing CIs that take into account fossil uncertainties. The posterior means of the root age ranged from 20 to 38 Myr among the five contigs. The posterior mean in the combined analysis is 33 Myr, with the 95% CI to be (29, 37).
Posterior Mean and 95% CI of Divergence Times (million years) Estimated from the Data of Steiper, Young, and Sukarna
Prior | A | B | C | D | E | All Concatenated | All Combined | |
|---|---|---|---|---|---|---|---|---|
| JC | ||||||||
| t1 (root) | 760 (86, 3074) | 28.2 (24.4, 32.2) | 30.1 (26.1, 34.5) | 34.6 (28.7, 41.5) | 35.2 (31.0, 39.9) | 38.1 (32.7, 44.3) | 33.1 (29.4, 37.1) | 32.3 (28.6, 36.2) |
| t2 (ape) | 7.0 (6.0, 8.0) | 6.9 (6.1, 7.8) | 5.9 (5.1, 6.7) | 6.6 (5.8, 7.6) | 5.8 (5.1, 6.5) | 6.6 (5.8, 7.5) | 5.9 (5.3, 6.6) | 5.8 (5.1, 6.5) |
| t3 (monkey) | 6.0 (5.0, 7.0) | 6.0 (5.3, 6.9) | 7.3 (6.4, 8.2) | 6.4 (5.5, 7.3) | 7.4 (6.5, 8.3) | 6.4 (5.6, 7.3) | 7.2 (6.4, 8.0) | 7.0 (6.2, 7.9) |
| r | 100 (12, 278) | 7.5 (6.5, 8.6) | 8.2 (7.1, 9.5) | 5.8 (4.8, 6.9) | 6.1 (5.4, 6.9) | 5.6 (4.8, 6.5) | 6.6 (5.8, 7.4) | |
| HKY + G5 | ||||||||
| t1 (root) | 760 (86, 3074) | 28.9 (25.1, 33.1) | 30.8 (26.7, 35.4) | 35.2 (29.2, 42.2) | 36.5 (32.1, 41.3) | 39.4 (33.7, 45.9) | 34.1 (30.3, 38.1) | 33.3 (29.5, 37.3) |
| t2 (ape) | 7.0 (6.0, 8.0) | 6.9 (6.1, 7.8) | 5.9 (5.1, 6.7) | 6.6 (5.8, 7.6) | 5.8 (5.1, 6.5) | 6.6 (5.8, 7.5) | 5.9 (5.3, 6.6) | 5.8 (5.2, 6.5) |
| t3 (monkey) | 6.0 (5.0, 7.0) | 6.0 (5.3, 6.9) | 7.3 (6.4, 8.2) | 6.4 (5.5, 7.3) | 7.4 (6.5, 8.3) | 6.4 (5.6, 7.3) | 7.2 (6.4, 8.0) | 7.0 (6.2, 7.9) |
| r | 1.00 (0.12, 2.78) | 7.5 (6.5, 8.6) | 8.3 (7.2, 9.5) | 5.8 (4.8, 6.9) | 6.1 (5.4, 6.9) | 5.6 (4.8, 6.6) | 6.6 (5.9, 7.4) | |
| κ | 3 (1.1, 5.8) | 4.17 (3.72, 4.66) | 3.73 (3.33, 4.17) | 3.31 (2.78, 3.91) | 4.35 (4.03, 4.69) | 3.86 (3.40, 4.37) | 4.05 (3.85, 4.25) | |
| α | 1 (.025, 3.69) | 1.01 (0.37, 2.55) | 1.34 (0.49, 3.31) | 1.66 (0.41, 4.40) | 0.61 (0.33, 1.13) | 0.86 (0.26, 2.43) | 0.77 (0.50, 1.18) |
Prior | A | B | C | D | E | All Concatenated | All Combined | |
|---|---|---|---|---|---|---|---|---|
| JC | ||||||||
| t1 (root) | 760 (86, 3074) | 28.2 (24.4, 32.2) | 30.1 (26.1, 34.5) | 34.6 (28.7, 41.5) | 35.2 (31.0, 39.9) | 38.1 (32.7, 44.3) | 33.1 (29.4, 37.1) | 32.3 (28.6, 36.2) |
| t2 (ape) | 7.0 (6.0, 8.0) | 6.9 (6.1, 7.8) | 5.9 (5.1, 6.7) | 6.6 (5.8, 7.6) | 5.8 (5.1, 6.5) | 6.6 (5.8, 7.5) | 5.9 (5.3, 6.6) | 5.8 (5.1, 6.5) |
| t3 (monkey) | 6.0 (5.0, 7.0) | 6.0 (5.3, 6.9) | 7.3 (6.4, 8.2) | 6.4 (5.5, 7.3) | 7.4 (6.5, 8.3) | 6.4 (5.6, 7.3) | 7.2 (6.4, 8.0) | 7.0 (6.2, 7.9) |
| r | 100 (12, 278) | 7.5 (6.5, 8.6) | 8.2 (7.1, 9.5) | 5.8 (4.8, 6.9) | 6.1 (5.4, 6.9) | 5.6 (4.8, 6.5) | 6.6 (5.8, 7.4) | |
| HKY + G5 | ||||||||
| t1 (root) | 760 (86, 3074) | 28.9 (25.1, 33.1) | 30.8 (26.7, 35.4) | 35.2 (29.2, 42.2) | 36.5 (32.1, 41.3) | 39.4 (33.7, 45.9) | 34.1 (30.3, 38.1) | 33.3 (29.5, 37.3) |
| t2 (ape) | 7.0 (6.0, 8.0) | 6.9 (6.1, 7.8) | 5.9 (5.1, 6.7) | 6.6 (5.8, 7.6) | 5.8 (5.1, 6.5) | 6.6 (5.8, 7.5) | 5.9 (5.3, 6.6) | 5.8 (5.2, 6.5) |
| t3 (monkey) | 6.0 (5.0, 7.0) | 6.0 (5.3, 6.9) | 7.3 (6.4, 8.2) | 6.4 (5.5, 7.3) | 7.4 (6.5, 8.3) | 6.4 (5.6, 7.3) | 7.2 (6.4, 8.0) | 7.0 (6.2, 7.9) |
| r | 1.00 (0.12, 2.78) | 7.5 (6.5, 8.6) | 8.3 (7.2, 9.5) | 5.8 (4.8, 6.9) | 6.1 (5.4, 6.9) | 5.6 (4.8, 6.6) | 6.6 (5.9, 7.4) | |
| κ | 3 (1.1, 5.8) | 4.17 (3.72, 4.66) | 3.73 (3.33, 4.17) | 3.31 (2.78, 3.91) | 4.35 (4.03, 4.69) | 3.86 (3.40, 4.37) | 4.05 (3.85, 4.25) | |
| α | 1 (.025, 3.69) | 1.01 (0.37, 2.55) | 1.34 (0.49, 3.31) | 1.66 (0.41, 4.40) | 0.61 (0.33, 1.13) | 0.86 (0.26, 2.43) | 0.77 (0.50, 1.18) |
NOTE.—Differences in rates and other substitution parameters were ignored in concatenated analysis and accounted for in combined analysis. Divergence times are defined in figure 7. Rate is ×10−10 substitutions per site per year.
Posterior Mean and 95% CI of Divergence Times (million years) Estimated from the Data of Steiper, Young, and Sukarna
Prior | A | B | C | D | E | All Concatenated | All Combined | |
|---|---|---|---|---|---|---|---|---|
| JC | ||||||||
| t1 (root) | 760 (86, 3074) | 28.2 (24.4, 32.2) | 30.1 (26.1, 34.5) | 34.6 (28.7, 41.5) | 35.2 (31.0, 39.9) | 38.1 (32.7, 44.3) | 33.1 (29.4, 37.1) | 32.3 (28.6, 36.2) |
| t2 (ape) | 7.0 (6.0, 8.0) | 6.9 (6.1, 7.8) | 5.9 (5.1, 6.7) | 6.6 (5.8, 7.6) | 5.8 (5.1, 6.5) | 6.6 (5.8, 7.5) | 5.9 (5.3, 6.6) | 5.8 (5.1, 6.5) |
| t3 (monkey) | 6.0 (5.0, 7.0) | 6.0 (5.3, 6.9) | 7.3 (6.4, 8.2) | 6.4 (5.5, 7.3) | 7.4 (6.5, 8.3) | 6.4 (5.6, 7.3) | 7.2 (6.4, 8.0) | 7.0 (6.2, 7.9) |
| r | 100 (12, 278) | 7.5 (6.5, 8.6) | 8.2 (7.1, 9.5) | 5.8 (4.8, 6.9) | 6.1 (5.4, 6.9) | 5.6 (4.8, 6.5) | 6.6 (5.8, 7.4) | |
| HKY + G5 | ||||||||
| t1 (root) | 760 (86, 3074) | 28.9 (25.1, 33.1) | 30.8 (26.7, 35.4) | 35.2 (29.2, 42.2) | 36.5 (32.1, 41.3) | 39.4 (33.7, 45.9) | 34.1 (30.3, 38.1) | 33.3 (29.5, 37.3) |
| t2 (ape) | 7.0 (6.0, 8.0) | 6.9 (6.1, 7.8) | 5.9 (5.1, 6.7) | 6.6 (5.8, 7.6) | 5.8 (5.1, 6.5) | 6.6 (5.8, 7.5) | 5.9 (5.3, 6.6) | 5.8 (5.2, 6.5) |
| t3 (monkey) | 6.0 (5.0, 7.0) | 6.0 (5.3, 6.9) | 7.3 (6.4, 8.2) | 6.4 (5.5, 7.3) | 7.4 (6.5, 8.3) | 6.4 (5.6, 7.3) | 7.2 (6.4, 8.0) | 7.0 (6.2, 7.9) |
| r | 1.00 (0.12, 2.78) | 7.5 (6.5, 8.6) | 8.3 (7.2, 9.5) | 5.8 (4.8, 6.9) | 6.1 (5.4, 6.9) | 5.6 (4.8, 6.6) | 6.6 (5.9, 7.4) | |
| κ | 3 (1.1, 5.8) | 4.17 (3.72, 4.66) | 3.73 (3.33, 4.17) | 3.31 (2.78, 3.91) | 4.35 (4.03, 4.69) | 3.86 (3.40, 4.37) | 4.05 (3.85, 4.25) | |
| α | 1 (.025, 3.69) | 1.01 (0.37, 2.55) | 1.34 (0.49, 3.31) | 1.66 (0.41, 4.40) | 0.61 (0.33, 1.13) | 0.86 (0.26, 2.43) | 0.77 (0.50, 1.18) |
Prior | A | B | C | D | E | All Concatenated | All Combined | |
|---|---|---|---|---|---|---|---|---|
| JC | ||||||||
| t1 (root) | 760 (86, 3074) | 28.2 (24.4, 32.2) | 30.1 (26.1, 34.5) | 34.6 (28.7, 41.5) | 35.2 (31.0, 39.9) | 38.1 (32.7, 44.3) | 33.1 (29.4, 37.1) | 32.3 (28.6, 36.2) |
| t2 (ape) | 7.0 (6.0, 8.0) | 6.9 (6.1, 7.8) | 5.9 (5.1, 6.7) | 6.6 (5.8, 7.6) | 5.8 (5.1, 6.5) | 6.6 (5.8, 7.5) | 5.9 (5.3, 6.6) | 5.8 (5.1, 6.5) |
| t3 (monkey) | 6.0 (5.0, 7.0) | 6.0 (5.3, 6.9) | 7.3 (6.4, 8.2) | 6.4 (5.5, 7.3) | 7.4 (6.5, 8.3) | 6.4 (5.6, 7.3) | 7.2 (6.4, 8.0) | 7.0 (6.2, 7.9) |
| r | 100 (12, 278) | 7.5 (6.5, 8.6) | 8.2 (7.1, 9.5) | 5.8 (4.8, 6.9) | 6.1 (5.4, 6.9) | 5.6 (4.8, 6.5) | 6.6 (5.8, 7.4) | |
| HKY + G5 | ||||||||
| t1 (root) | 760 (86, 3074) | 28.9 (25.1, 33.1) | 30.8 (26.7, 35.4) | 35.2 (29.2, 42.2) | 36.5 (32.1, 41.3) | 39.4 (33.7, 45.9) | 34.1 (30.3, 38.1) | 33.3 (29.5, 37.3) |
| t2 (ape) | 7.0 (6.0, 8.0) | 6.9 (6.1, 7.8) | 5.9 (5.1, 6.7) | 6.6 (5.8, 7.6) | 5.8 (5.1, 6.5) | 6.6 (5.8, 7.5) | 5.9 (5.3, 6.6) | 5.8 (5.2, 6.5) |
| t3 (monkey) | 6.0 (5.0, 7.0) | 6.0 (5.3, 6.9) | 7.3 (6.4, 8.2) | 6.4 (5.5, 7.3) | 7.4 (6.5, 8.3) | 6.4 (5.6, 7.3) | 7.2 (6.4, 8.0) | 7.0 (6.2, 7.9) |
| r | 1.00 (0.12, 2.78) | 7.5 (6.5, 8.6) | 8.3 (7.2, 9.5) | 5.8 (4.8, 6.9) | 6.1 (5.4, 6.9) | 5.6 (4.8, 6.6) | 6.6 (5.9, 7.4) | |
| κ | 3 (1.1, 5.8) | 4.17 (3.72, 4.66) | 3.73 (3.33, 4.17) | 3.31 (2.78, 3.91) | 4.35 (4.03, 4.69) | 3.86 (3.40, 4.37) | 4.05 (3.85, 4.25) | |
| α | 1 (.025, 3.69) | 1.01 (0.37, 2.55) | 1.34 (0.49, 3.31) | 1.66 (0.41, 4.40) | 0.61 (0.33, 1.13) | 0.86 (0.26, 2.43) | 0.77 (0.50, 1.18) |
NOTE.—Differences in rates and other substitution parameters were ignored in concatenated analysis and accounted for in combined analysis. Divergence times are defined in figure 7. Rate is ×10−10 substitutions per site per year.
As discussed earlier, in the limit of an infinite number of sites, the marginal distribution of each divergence time should be a simple transformation of the posterior density of r. In that case, the width of the confidence interval for each divergence time estimate should asymptotically become a linear function of the mean of the posterior. Thus, a simple way to examine the amount of information in the sequence data is to regress the mean against the width of the confidence interval for each node. Plotting the posterior CI bounds against the posterior means of divergence times for the data of Steiper, Young, and Sukarna (2004) revealed a nearly perfect linear relationship (results not shown). While there are only three time estimates, we suspect that the amount of sequence data has nearly saturated, and adding more sites is unlikely to improve the precision of posterior time estimation (compare with the simulation results above). The 95% CI width had a regression coefficient of 0.23 against the posterior mean, meaning that every million years of species divergence adds 0.23 Myr to the 95% CI width.
The two substitution models JC and HKY + G produced very similar results, despite the fact that HKY + G fits the data far better than JC (results not shown). For the two calibration nodes, the posterior means and 95% CIs are identical between the two models at this level of accuracy. Estimates of rates under the two models were also identical. This lack of difference between the two models appears partly due to the high similarities of the sequences.
We examined the effect of the prior for divergence times on posterior time estimation, using the HKY + G model for combined analysis of all five contigs. The birth-death prior with λ = μ = 2 and ρ = 0.1, used above (table 2), specifies a nearly flat kernel density between 0 and t1 = 1 for node ages t−1. We used two additional sets of parameters to explore the effect of prior tree shape. In the second set, λ = 1, μ = 10, and ρ = 0.1, and the kernel density has a highly skewed L shape, meaning that the nonroot internal nodes tend to be near the tips with long internal branches in the tree. In the third set, λ = 10, μ = 1, and ρ = 10−4, which produces an inverse L shaped kernel density, favoring starlike trees. The second set of prior parameters led to the posterior mean 32.8 Myr with the 95% CI to be (29.2, 36.8) for the root age t1. The estimates are similar to those of table 2 and are only slightly younger. The posterior estimates of ages for the two calibration nodes were younger as well, but the differences were very small; for example, the human-chimpanzee divergence was dated to 5.7 Myr (5.1, 6.4). Under the third prior, the posterior mean of t1 was 35 Myr with the 95% CI to be (31.1, 39.3). The ages were slightly older than those of table 2. Overall, we found that parameters λ, μ, and ρ had only minor effects on posterior time estimates.
We also changed the gamma priors for the two fossil node ages into a uniform distribution with soft lower and upper bounds, that is, “>0.06 < 0.08” for the human-chimpanzee divergence and “>0.05 < 0.07” for the baboon-macaque divergence. The posterior means and 95% CIs became 33.7 Myr (32.1, 35.4) for t1, 6.0 Myr (5.8, 6.3) for t2, and 7.0 Myr (6.7, 7.2) for t3. The posterior means were virtually identical to those under the gamma priors (table 2, “All combined, HKY + G”), but the CIs were narrower. If we assume more uncertainty in the fossil dates, with bounds “>0.05 < 0.09” for the human-chimpanzee divergence and “>0.05 < 0.08” for the baboon-macaque divergence, the posterior estimates became 32.7 Myr (28.0, 37.9) for t1, 5.6 Myr (4.9, 6.5) for t2, and 7.0 Myr (5.9, 8.0) for t3. The posterior means did not change much, but the CIs all became much wider, as expected.
The Mitochondrial Data Set of Cao et al.
This data set consists of all 12 protein-coding genes encoded by the same strand of the mitochondrial genome from seven species of apes (Cao et al. 1998). The species are human (H. sapiens), common chimpanzee (P. troglodytes), bonobo (Pan paniscus), gorilla (Gorilla gorilla), Bornean orangutan (Pongo pygmaeus pygmaeus), Sumatran orangutan (Pongo pygmaeus abelii), and common gibbon (Hylobates lar). The 12 protein-coding genes are concatenated into one supergene and analyzed as one data set as they appear to have similar substitution patterns. Instead, we accommodate the large differences among the three codon positions. After removal of sites with alignment gaps, the sequence has 3,331 nt sites at each codon position. See Cao et al. (1998) for the GenBank accession numbers.
The species phylogeny is shown in figure 1. Two fossil calibrations were used in our Bayesian analysis. The first is for the human-chimpanzee divergence, assumed to be between 6 and 8 Myr, with a most likely date of 7 Myr. A gamma prior G(186.2, 2672.6) is used for the node age, as in the previous data set. The second calibration is for the divergence of the orangutan from the African apes, assumed to be between 12 and 16 Myr, with a most likely date of 14 Myr (Raaum et al. 2005). The prior is specified as “>0.12 = 0.139 < 0.16,” and the gamma G(186.9, 1337.7) is fitted, with tail probabilities 2.2% and 2.7%.
We analyze the three codon positions separately and then combined. Table 3 lists posterior means and 95% CIs for divergence times, substitution rates, and substitution parameters κ and α. The estimates for the three codon positions were in the order r2 < r1 < r3, κ2 < κ1 < κ3, and α2 < α1 < α3, consistent with well-known patterns of conserved evolution of the mitochondrial genes (see, e.g., Kumar 1996). In the combined analysis, posterior estimates of rates rs, κs, and αs (not shown) are very similar to those obtained in the separate analyses (table 3).
Posterior Mean and 95% CIs of Divergence Times (million years) for the Mitochondrial Data of Cao et al.
Prior | Position pos1, HKY + G | Position pos2, HKY + G | Position pos3, HKY + G | All, HKY + G | All, JC | |
|---|---|---|---|---|---|---|
| t1 (root) | 1376 (268, 4857) | 17.8 (15.3, 20.6) | 17.4 (14.7, 20.9) | 22.6 (19.6, 26.0) | 19.8 (17.5, 22.2) | 15.0 (13.4, 16.7) |
| t2 | 14.0 (12.0, 16.1) | 15.7 (13.9, 17.6) | 15.3 (13.5, 17.3) | 16.3 (14.5, 18.1) | 16.3 (14.6, 18.1) | 14.0 (12.6, 15.5) |
| t3 | 10.5 (6.9, 14.5) | 8.5 (7.3, 9.8) | 9.0 (7.5, 10.8) | 8.6 (7.6, 9.8) | 8.6 (7.6, 9.6) | 9.4 (8.5, 10.5) |
| t4 (HC) | 7.0 (6.0, 8.0) | 6.3 (5.5, 7.1) | 6.5 (5.6, 7.3) | 6.2 (5.5, 6.9) | 6.1 (5.5, 6.8) | 7.1 (6.4, 7.9) |
| t5 | 3.5 (0.2, 7.0) | 2.7 (2.1, 3.4) | 2.4 (1.5, 3.5) | 1.9 (1.6, 2.2) | 2.0 (1.8, 2.4) | 2.9 (2.6, 3.3) |
| t6 | 7.0 (0.3, 14.0) | 4.8 (3.8, 5.8) | 4.8 (3.6, 6.2) | 3.7 (3.1, 4.3) | 4.1 (3.5, 4.7) | 5.2 (4.6, 5.9) |
| r | 1.00 (0.12, 2.78) | 0.492 (0.423, 0.571) | 0.177 (0.146, 0.212) | 3.21 (2.81, 3.68) | ||
| κ | 3 (1.1, 5.8) | 12.3 (10.4, 14.4) | 9.3 (7.3, 11.8) | 34.7 (30.9, 38.9) | ||
| α | 1 (0.025, 3.69) | 0.225 (0.180, 0.279) | 0. 047 (0. 004, 0.115) | 3.71 (2.64, 5.31) |
Prior | Position pos1, HKY + G | Position pos2, HKY + G | Position pos3, HKY + G | All, HKY + G | All, JC | |
|---|---|---|---|---|---|---|
| t1 (root) | 1376 (268, 4857) | 17.8 (15.3, 20.6) | 17.4 (14.7, 20.9) | 22.6 (19.6, 26.0) | 19.8 (17.5, 22.2) | 15.0 (13.4, 16.7) |
| t2 | 14.0 (12.0, 16.1) | 15.7 (13.9, 17.6) | 15.3 (13.5, 17.3) | 16.3 (14.5, 18.1) | 16.3 (14.6, 18.1) | 14.0 (12.6, 15.5) |
| t3 | 10.5 (6.9, 14.5) | 8.5 (7.3, 9.8) | 9.0 (7.5, 10.8) | 8.6 (7.6, 9.8) | 8.6 (7.6, 9.6) | 9.4 (8.5, 10.5) |
| t4 (HC) | 7.0 (6.0, 8.0) | 6.3 (5.5, 7.1) | 6.5 (5.6, 7.3) | 6.2 (5.5, 6.9) | 6.1 (5.5, 6.8) | 7.1 (6.4, 7.9) |
| t5 | 3.5 (0.2, 7.0) | 2.7 (2.1, 3.4) | 2.4 (1.5, 3.5) | 1.9 (1.6, 2.2) | 2.0 (1.8, 2.4) | 2.9 (2.6, 3.3) |
| t6 | 7.0 (0.3, 14.0) | 4.8 (3.8, 5.8) | 4.8 (3.6, 6.2) | 3.7 (3.1, 4.3) | 4.1 (3.5, 4.7) | 5.2 (4.6, 5.9) |
| r | 1.00 (0.12, 2.78) | 0.492 (0.423, 0.571) | 0.177 (0.146, 0.212) | 3.21 (2.81, 3.68) | ||
| κ | 3 (1.1, 5.8) | 12.3 (10.4, 14.4) | 9.3 (7.3, 11.8) | 34.7 (30.9, 38.9) | ||
| α | 1 (0.025, 3.69) | 0.225 (0.180, 0.279) | 0. 047 (0. 004, 0.115) | 3.71 (2.64, 5.31) |
NOTE.—Divergence times are defined in figure 1. Rate is measured by the number of substitutions per 108 years. HC indicates time human-chimp divergence.
Posterior Mean and 95% CIs of Divergence Times (million years) for the Mitochondrial Data of Cao et al.
Prior | Position pos1, HKY + G | Position pos2, HKY + G | Position pos3, HKY + G | All, HKY + G | All, JC | |
|---|---|---|---|---|---|---|
| t1 (root) | 1376 (268, 4857) | 17.8 (15.3, 20.6) | 17.4 (14.7, 20.9) | 22.6 (19.6, 26.0) | 19.8 (17.5, 22.2) | 15.0 (13.4, 16.7) |
| t2 | 14.0 (12.0, 16.1) | 15.7 (13.9, 17.6) | 15.3 (13.5, 17.3) | 16.3 (14.5, 18.1) | 16.3 (14.6, 18.1) | 14.0 (12.6, 15.5) |
| t3 | 10.5 (6.9, 14.5) | 8.5 (7.3, 9.8) | 9.0 (7.5, 10.8) | 8.6 (7.6, 9.8) | 8.6 (7.6, 9.6) | 9.4 (8.5, 10.5) |
| t4 (HC) | 7.0 (6.0, 8.0) | 6.3 (5.5, 7.1) | 6.5 (5.6, 7.3) | 6.2 (5.5, 6.9) | 6.1 (5.5, 6.8) | 7.1 (6.4, 7.9) |
| t5 | 3.5 (0.2, 7.0) | 2.7 (2.1, 3.4) | 2.4 (1.5, 3.5) | 1.9 (1.6, 2.2) | 2.0 (1.8, 2.4) | 2.9 (2.6, 3.3) |
| t6 | 7.0 (0.3, 14.0) | 4.8 (3.8, 5.8) | 4.8 (3.6, 6.2) | 3.7 (3.1, 4.3) | 4.1 (3.5, 4.7) | 5.2 (4.6, 5.9) |
| r | 1.00 (0.12, 2.78) | 0.492 (0.423, 0.571) | 0.177 (0.146, 0.212) | 3.21 (2.81, 3.68) | ||
| κ | 3 (1.1, 5.8) | 12.3 (10.4, 14.4) | 9.3 (7.3, 11.8) | 34.7 (30.9, 38.9) | ||
| α | 1 (0.025, 3.69) | 0.225 (0.180, 0.279) | 0. 047 (0. 004, 0.115) | 3.71 (2.64, 5.31) |
Prior | Position pos1, HKY + G | Position pos2, HKY + G | Position pos3, HKY + G | All, HKY + G | All, JC | |
|---|---|---|---|---|---|---|
| t1 (root) | 1376 (268, 4857) | 17.8 (15.3, 20.6) | 17.4 (14.7, 20.9) | 22.6 (19.6, 26.0) | 19.8 (17.5, 22.2) | 15.0 (13.4, 16.7) |
| t2 | 14.0 (12.0, 16.1) | 15.7 (13.9, 17.6) | 15.3 (13.5, 17.3) | 16.3 (14.5, 18.1) | 16.3 (14.6, 18.1) | 14.0 (12.6, 15.5) |
| t3 | 10.5 (6.9, 14.5) | 8.5 (7.3, 9.8) | 9.0 (7.5, 10.8) | 8.6 (7.6, 9.8) | 8.6 (7.6, 9.6) | 9.4 (8.5, 10.5) |
| t4 (HC) | 7.0 (6.0, 8.0) | 6.3 (5.5, 7.1) | 6.5 (5.6, 7.3) | 6.2 (5.5, 6.9) | 6.1 (5.5, 6.8) | 7.1 (6.4, 7.9) |
| t5 | 3.5 (0.2, 7.0) | 2.7 (2.1, 3.4) | 2.4 (1.5, 3.5) | 1.9 (1.6, 2.2) | 2.0 (1.8, 2.4) | 2.9 (2.6, 3.3) |
| t6 | 7.0 (0.3, 14.0) | 4.8 (3.8, 5.8) | 4.8 (3.6, 6.2) | 3.7 (3.1, 4.3) | 4.1 (3.5, 4.7) | 5.2 (4.6, 5.9) |
| r | 1.00 (0.12, 2.78) | 0.492 (0.423, 0.571) | 0.177 (0.146, 0.212) | 3.21 (2.81, 3.68) | ||
| κ | 3 (1.1, 5.8) | 12.3 (10.4, 14.4) | 9.3 (7.3, 11.8) | 34.7 (30.9, 38.9) | ||
| α | 1 (0.025, 3.69) | 0.225 (0.180, 0.279) | 0. 047 (0. 004, 0.115) | 3.71 (2.64, 5.31) |
NOTE.—Divergence times are defined in figure 1. Rate is measured by the number of substitutions per 108 years. HC indicates time human-chimp divergence.
Estimates of divergence times were similar at the three codon positions, except for the age of the root t1, for which the first two positions produced younger estimates (∼18 Myr) than the third (∼23 Myr). The posterior mean of t1 from the combined data was 20 Myr with the 95% CI (19 and 26 Myr). For the human-chimpanzee divergence (t4), the estimated age from the combined analysis was 6.1 Myr (5.5, 6.8). In figure 8, the widths (w) of the 95% CIs were plotted against the posterior means of node ages (t). For the combined analysis, the six points were nearly perfectly linearly related, suggesting that the amount of data had nearly reached saturation, and increasing the sequence length was unlikely to improve the precision of time estimates. The regression line w = 0.23t means that even with infinitely many sites in the sequence every 1 Myr of species divergence would add 0.23 Myr to the 95% CI. In the separate analyses of the three codon positions, the linear fit was better at the third position and poorer at the second, indicating that the third positions were more variable and informative than the second (fig. 8).
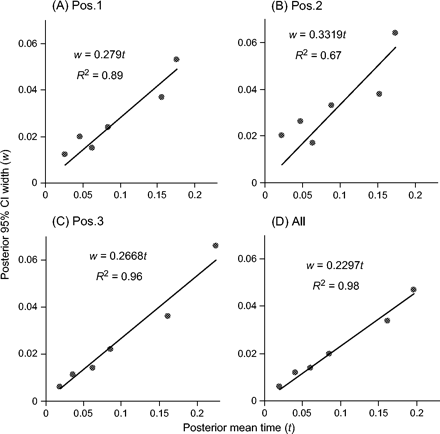
The widths of the posterior 95% CIs plotted against the posterior means of the divergence times in separate analyses of the three codon positions and in combined analysis of the mitochondrial data set. The six divergence times are shown in figure 1.
The JC model gave somewhat younger estimates for the root age compared with the corresponding estimates under HKY + G. The estimates from the combined analysis under JC were shown in table 3, where the posterior mean for the root age was 15 Myr, with the 95% CI to be (13 and 17 Myr). The JC model is ineffective at correcting for multiple hits and is expected to be unreliable for these data.
We again examined the effect of the prior for divergence times on posterior time estimation, using two alternative sets of λ, μ, and ρ in the birth-death process model. The HKY + G model is used for combined analysis of all three codon positions, in comparison with results of table 3 (“All, HKY + G”). Under the second prior (λ = 1, μ = 10, and ρ = 0.1), the kernel density is L shaped. Posterior time estimates all became slightly younger, but the effects were small. For example, the root age had the posterior mean 19.3 Myr with the 95% CI (17.1, 21.8), slightly younger than 19.8 Myr (17.5, 22.3) (table 3). Under the third set (λ = 10, μ = 1, and ρ = 10−4), the kernel density has an inverse L shape, favoring starlike trees. The posterior mean for the root age became 20.5 Myr (18.1, 23.1). The ages were slightly older than those of table 3. Similarly, all other divergence times became slightly older with this prior. The patterns were the same as in the previous data set. Overall, the different priors on divergence times produced very similar posterior time estimates in this data set.
We also changed the gamma priors for the two fossil node ages into uniform priors with soft lower and upper bounds: “>0.06 < 0.08” for the human-chimpanzee divergence and “>0.12 < 0.16” for the orangutan divergence. With this prior, the posterior means and 95% CIs became 19.3 Myr (17.9, 20.8) for the root age t1, and 6.1 Myr (5.8, 6.4) for the human-chimpanzee divergence t4. The posterior means were very similar to those under the gamma priors (table 3 “All, HKY + G”), but the CIs were narrower. If we use looser bounds to allow more uncertain fossil dates, that is, “>0.05 < 0.09” for the human-chimpanzee divergence and “>0.11 < 0.18” for the orangutan divergence, the posterior means and 95% CIs became 20.2 Myr (17.0, 22.7) for root age t1 and 6.0 Myr (5.0, 6.8) for the human-chimpanzee divergence. The posterior means did not change much, but all the CIs became much wider, as expected. The patterns were the same as in the previous data set.
Discussion
We note that our strategy of specifying priors for divergence times would work if a different kernel density is used in place of equation (4). The theory of order statistics can then be used to incorporate arbitrary densities to describe uncertainties in fossil dates, as in this paper. We prefer the birth-death process with species sampling as it has a biological interpretation. With three parameters (λ, μ, and ρ), the model can generate different tree shapes as reflected in the relative node ages and is sufficiently flexible to accommodate various data sets. In particular, the sampling fraction was noted to dramatically affect the shape of the tree (Yang and Rannala 1997). While small trees may not contain enough information to reliably estimate those parameters, we suggest that varying them to change the tree shape in the prior provides a convenient way of assessing the robustness of the posterior distribution of divergence times to the prior specifications.
We also suggest that it is important to explore the sensitivity of posterior time estimates to the specification of fossil calibration priors. Results obtained from both our simulation study and from analyzing the two real data sets demonstrate the critical importance of reliable high-precision fossil calibrations. It does not seem to be sufficiently appreciated that increasing the amount of sequence data cannot be expected to reduce errors in time estimates to zero. Both our theoretical analysis of the normal distribution example and our simulations using good fossils demonstrate that the posterior confidence intervals will typically be comparable in width to the most precise prior interval even if infinitely many sites are in the sequence. For readers dismayed by such results, we offer the consolation that the problem will become much worse when the molecular clock is relaxed. We note that Bayesian estimation using hard bounds sometimes produced very narrow posterior CIs because age estimates converge to the prior bounds. Based on these results, we suggest that exceptionally narrow confidence intervals may often not represent genuinely high precision in posterior divergence time estimates but rather conflicts among fossil calibrations or conflicts between fossils and sequences. We suggest that soft bounds are in general preferred to hard bounds for describing fossil uncertainties. The risk of using zero probabilities in a prior is well known in statistics, characterized by the Bayesian statistician D. V. Lindley as the Cromwell's rule: one should avoid using prior probabilities of 0 (or 1). Such extreme priors force the posterior probabilities to be 0 (or 1) as well, whatever be the data. Oliver Cromwell famously wrote to the synod of the Church of Scotland on August 5, 1650, saying “I beseech you, in the bowels of Christ, think it possible you may be mistaken.” As Lindley (1985, p. 104) puts it, if you attach a prior probability of 0 to the hypothesis that the moon is made of green cheese, then even whole armies of astronauts returning from the moon bearing green cheese cannot convince you.
Implementation Details and Program Availability
The method developed in this paper is implemented in the MCMCTREE program in the PAML package (Yang 1997). Fossil information is specified as part of the tree notation, with “>” and “<” indicating lower and upper bounds, respectively. In addition, if a most likely age is specified using “=,” a gamma distribution is fitted to the node age. For example, the tree notation “((human, chimp) >0.06 = 0.0693 < 0.08, (baboon, rhesus macaque) > 0.05 < 0.07)” specifies that the human-chimpanzee divergence was between 6 and 8 Myr, with a most likely age at 6.93 Myr. The program will then fit a gamma distribution. The baboon-macaque divergence is between 5 and 7 Myr, and the program will use soft lower and upper bounds. Here time is measured in 100 Myr.
Appendix A. Proposal Steps in the MCMC
The MCMC algorithm implemented in this paper involves four proposal steps, each of which updates some parameters in the Markov chain. These steps are described briefly below. The reader is referred to earlier papers in the area, such as Thorne, Kishino, and Painter (1998), Drummond et al. (2002), and Rannala and Yang (2003), for more detailed discussions of Bayesian MCMC algorithms. Each of the four steps below involves a fine-tuning parameter that acts as the step length. Larger steps usually lead to rejection of most proposals, while small steps lead to high acceptance proportions. The fine-tuning parameters should be adjusted to achieve intermediate acceptance proportions, say, between 10% and 80%.
Step 1. Updating Divergence Times at Internal Nodes of the Species Tree
This step cycles through the internal nodes in the species tree to propose changes to the node ages (divergence times). The node age is bounded by the ages of the daughter nodes and the mother node. A sliding window is used to propose the new age.
Step 2. Updating Substitution Rates at Different Loci
For each locus, the current rate is multiplied by a random variable around 1; that is, the current rate is expanded or shrunk by a random constant.
Step 3. Updating Substitution Parameters in the Model
This step is used only if there are parameters in the substitution model, such as κ and α under HKY + G. It is not used under JC. The step cycles through all the loci and, for each locus, updates the parameter by multiplying the current value with a random variable around 1.
Step 4. Mixing Step
This step is similar to the mixing step of Thorne, Kishino, and Painter (1998) and Rannala and Yang (2003). We generate a random variable around 1: c = eε(r − 0.5), where r ∼ U(0, 1) and ε is a small constant. We then multiply all (s − 1) divergence times by c and divide all g rates at the g loci by c. The proposal ratio is c(s − 1) − g. As the branch lengths are not changed, there is no need to update the likelihood.
Appendix B. Identifiability Problem in a Normal Distribution Example
Pekka Pamilo, Associate Editor
We thank Adrian Friday, Michael E. Steiper, Anne Yoder, and two anonymous referees for many constructive comments. This study is supported by a grant from the Natural Environment Research Council (United Kingdom) to Z.Y. and National Institutes of Health grant HG01988 to B.R.
References
Aris-Brosou, S., and Z. Yang.
———.
Brunet, M., F. Guy, D. Pilbeam et al. (37 co-authors).
Cao, Y., A. Janke, P. J. Waddell, M. Westerman, O. Takenaka, S. Murata, N. Okada, S. Paabo, and M. Hasegawa.
Delson, E., I. Tattersall, J. A. Van Couvering, and A. S. Brooks. ed.
Drummond, A. J., G. K. Nicholls, A. G. Rodrigo, and W. Solomon.
Felsenstein, J.
Graur, D., and W. Martin.
Hasegawa, M., H. Kishino, and T. Yano.
Hastings, W. K.
Ho, S. Y. W., M. J. Phillips, A. J. Drummond, and A. Cooper.
Jukes, T. H., and C. R. Cantor.
Kishino, H., J. L. Thorne, and W. J. Bruno.
Kumar, S.
Metropolis, N., A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, and E. Teller.
Raaum, R. L., K. N. Sterner, C. M. Noviello, C. B. Stewart, and T. R. Disotell.
Rambaut, A., and L. Bromham.
Rannala, B., and Z. Yang.
———.
Sanderson, M. J.
Steiper, M. E., N. M. Young, and T. Y. Sukarna.
Suchard, M. A., R. E. Weiss, and J. S. Sinsheimer.
Thorne, J. L., H. Kishino, and I. S. Painter.
Yang, Z.
———.
———.
———.
Yang, Z., R. Nielsen, and M. Hasegawa.
Yang, Z., and B. Rannala.
Yoder, A. D., and Z. Yang.
Author notes
*Department of Biology, University College London, London, United Kingdom; and †Genome Center, University of California, Davis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}